GCK has the highest Km (lowest affinity) for glucose among all canonical HKs (HK1-4), allowing the liver and pancreas to serve as a “glucose buffer” and a “glucose sensor,” respectively. It is not inhibited by G6P and has a 50-fold lower affinity for glucose than other isoforms. Within the liver, the low affinity is tailored to ensure the availability of glucose to physiologically sensitive tissues such as the brain under starvation and its utilization only when glucose is abundantly available. Within the pancreas, this feature allows GCK to act as a “
glucose sensor” to regulate insulin release. Mutations in the glucokinase (GCK) gene lead to maturity-onset diabetes of the young, type 2 (MODY-2), and persistent hyperinsulinemic hypoglycemia of infancy (PHHI)
[9][10][11][47,49,83]. MODY-2 is a mild type 2 diabetes resulting from a defect in glucose-induced insulin secretion
[9][10][12][47,48,49]. Mutations in the GCK leading to MODY-2 are arguably the most common cause of monogenic diabetes due to these specific mutations. More than 40 mutations have been linked to MODY-2, including frameshifts, nonsense, missense, and splice-site variants
[1][2][3][13][14][15][16][1,2,3,4,5,6,7]. The proposed role of GCK as a “glucose sensor” in pancreatic β-cells
[5][14][17][2,11,12] is consistent with the MODY-2 phenotype wherein small reductions in β-cell activity increase the threshold for glucose-induced insulin secretion resulting in the phenotype. However, a report by Postic et al. suggests that hepatic GCK also plays a role in MODY-2. Alterations in GCK activity are also associated with many other diseases that have been reviewed elsewhere in detail
[17][18][12,42]. Owing to its unique role, GCK regulation is complex, and several regulatory mechanisms have been discovered. Alternative and tissue-specific promoters drive GCK transcription and gene expression to varying degrees
[19][20][21][22][23][24][84,85,86,87,88,89]. Several metabolites, including insulin, glucose, and hormones, regulate GCK expression at the transcriptional level
[25][26][27][28][29][43,90,91,92,93]. The regulation of GCK has been recently reviewed elsewhere in more detail
[30][94].
2. Roles of Hexokinases in Cancer-Mediated Metabolic Reprogramming
One of the characteristic features of cancer is unabated cell division. For this reason, neoplastic cells preferentially obtain energy and biomolecules through glycolysis through metabolic reprogramming. Metabolic reprogramming refers to the ability of cancer cells to alter their metabolism to support their enhanced metabolic requirements of high ATP and intermediates for biosynthetic processes. This requirement brings about extensive changes in the expression of different hexokinase enzymes.
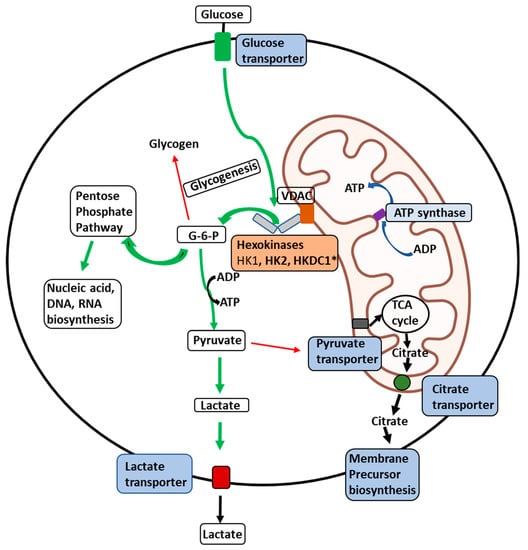
Hexokinase 1: The expression of HK1 is amplified in some cancers where it is responsible for rewiring the metabolic state towards aerobic glycolysis to supply ATP and macromolecules (
Figure 1)
[34][35][36][103,104,105]. The observation that most normal cells express HK1 while cancer cells express HK1 and HK2 stimulated interest in reducing HK2 activity in cancers. However, studies have demonstrated that the knockdown of HK2 alone does not inhibit in vivo tumor progression with reduced glucose consumption, suggesting that HK1 compensates for the overall tumorigenic potential. In contrast, the knockdown of HK2 in HK1- HK2+ cancers reduced xenograft tumor progression
[37][38][39][40][106,107,108,109]. These studies suggest a greater involvement of HK1 in tumor progression beyond its currently known role and possibly as a regulatory function in cancer cells. For example, in a study by Daniela et al., HK1 has been shown to be involved in ovarian cancer in a glucose phosphorylation-independent fashion
[41][110]. HK1 also serves as the effector of KRAS4A, an isoform of the most frequently mutated oncogene KRAS, during tumorigenesis
[42][111].
Figure 1. Illustration of the delivery of glucose to membrane-bound HKs in malignant cells. Illustration of the glucose delivery to HKs 1, 2, and HKDC1 bound to the outer mitochondrial membrane (OMM) and metabolic fates of the glucose-6-phosphate (G6P) formed thereof within a malignant cell. Glucose transport across the plasma membrane by glucose transporters is phosphorylated by HKs (HK1, HK2, or HKDC1) bound to a voltage-dependent anion channel (VDAC) located on the outer mitochondrial membrane. VDAC allows direct access of ATP generated by the ATP synthase within the mitochondria to the HKs, which can be transported across the inner-mitochondrial membrane by the adenine nucleotide translocator. To maintain malignant cells’ highly glycolytic metabolic flux, the product G6P is rapidly distributed across key metabolic routes (see thick green arrows). The primary metabolic routes for G6P are either entry into the pentose-phosphate pathway for biosynthesis of nucleic acid precursors or conversion to pyruvate and lactate through glycolysis. In cancer cells, most lactate is transported out of the cells with the aid of lactate transporters. In contrast, small amounts of pyruvate are transported to mitochondria through the pyruvate transporters to supply intermediates to the tricarboxylic acid (TCA) cycle (thin red arrows). Citrate transporters transport citrate produced in the TCA cycle to aid in synthesizing membrane components such as phospholipids and cholesterol, essential for tumor cell proliferation. * Novel hexokinase, HKDC1, with roles still unknown.
Hexokinase 2: HK2 is significantly overexpressed in treatment-resistant primary and metastatic breast cancer
[43][44][45][46][37,38,39,40], bladder cancer
[47][112], cervical squamous cell carcinoma
[48][113], colorectal cancer
[49][114], neuroendocrine tumor
[34][103], ovarian epithelial tumors
[35][104], glioblastoma
[36][50][55,105], hepatocellular carcinoma
[51][30], laryngeal squamous cell carcinoma
[52][31], lung cancer
[53][32], neuroblastoma
[54][33], pancreatic cancer
[55][34], and prostate cancer. HK2 expression in these cancers inversely correlates to overall patient survival rates
[56][35]. The genetic ablation of HK2 is known to inhibit malignant growth in mouse models
[37][38][39][40][57][36,106,107,108,109]. A landmark study on an adult tumor model of mice demonstrated the therapeutic effects of systemic deletion of HK2
[57][58][59][60][61][36,115,116,117,118]. In addition to its enzymatic activity, the mitochondrial binding ability of HK2 plays a role in inhibiting apoptosis and upregulating synthetic pathways which support tumor growth (
Figure 1). The mitochondrial-bound HK2 is therefore elevated in many forms of cancer
[43][44][45][37,38,39]. The amplification of HK2 appears to be related to the expression of p53. Recent studies have shown that p53-inducible protein TIGAR (Tp53-induced glycolysis and apoptosis regulator), Akt, and ER stress sensor kinase could regulate mitochondrial HK2 localization
[53][60][62][63][64][65][66][32,117,119,120,121,122,123]. Interestingly, the mitochondrial TIGAR–HK2 complex upregulated HK2 and hypoxia-inducible factor 1 (HIF1) activity, which limits reactive oxygen species (ROS) production and protects against tumor cell death under hypoxic conditions
[67][68][69][70][71][72][124,125,126,127,128,129]. It is also observed that the GCK to HK2 switch occurs in hepatocellular carcinoma (HCC), and the expression of HK2 is highest in HCC
[70][127]. Additionally, HK2 is also regulated by epigenetic mediators, including long non-coding RNAs
[37][38][44][45][38,39,106,107], microRNAs
[66][67][68][69][70][123,124,125,126,127], histone, and DNA methylation
[40][109]. HK2 is localized to the outer mitochondrial membrane through a voltage-dependent anion channel (VDAC)
[69][126] (
Figure 1). This association permits direct access to the ATP generated within the mitochondria
[67][124]. This phenomenon is especially significant in malignant cells where rates of aerobic glycolysis go up tremendously to meet the energy demands of the transformed cell (Warburg effect)
[36][105].
Hexokinase 3: HK3 is upregulated in several cancers, including acute myeloid leukemia (AML), where it plays the role of an anti-apoptotic protein to promote tumor cell survival alongside HK1 and 2
[73][74][130,131]. The previously identified functions of the enzyme include cell survival through the attenuation of apoptosis and the enhancement of mitochondrial biogenesis
[14][75][76][2,132,133]. The latest research about the functions of HK3 in normal and cancer cells has uncovered previously unanticipated roles of this protein. A recent study by Seiler et al. has reported that hexokinase 3 enhances myeloid cell survival via non-glycolytic functions
[77][134]. In contrast, another report by Xu et al. showed that HK3 dysfunction promotes tumorigenesis and immune escape by upregulating macrophage infiltration in renal cell carcinoma
[78][135].
Glucokinase: Glucose phosphorylation activity for GCK has been observed in several cancer cell lines
[79][136]. GCK is also known to interact with BAD (Bcl-2 agonist of cell death) to integrate glycolysis with apoptosis
[80][81][82][83][50,137,138,139]. To date, 17 activating mutations targeted by multiple activators have been identified in the allosteric activator site of GCK
[84][85][86][87][140,141,142,143]. The activating variations and their targeting by the activators lead to enhanced cellular proliferation, including the proliferation of cancer cell lines such as INS, which indicates a putative pro-oncogenic role for GCK
[88][89][90][144,145,146]. Although there is no direct evidence for the role of GCK as a pro-oncogene, recent reports exploring somatic variations of allosterically regulated proteins in cancer genomes suggest that somatic mutations of GCK could play a role in tumorigenesis
[91][147]. Těšínský et al. provide the first direct evidence of the role of GCK in tumorigenesis by demonstrating a change in the kinetic properties of GCK which include an increased affinity for glucose and changes in cooperative binding
[92][148].
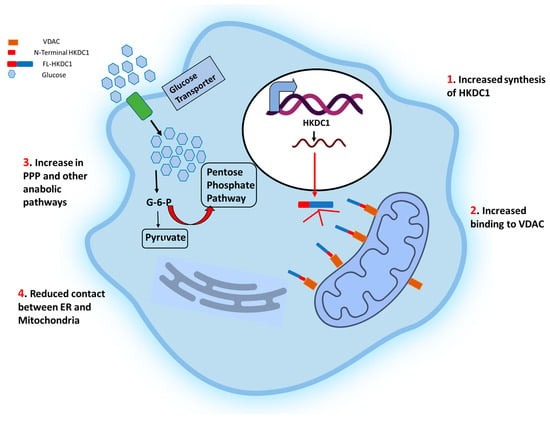
Hexokinase domain containing 1: Studies performed over the past decade have linked HKDC1 to various functions (
Figure 2). Much of the interest in HKDC1’s role in cancer stems from the fact that, like HK1 and 2, it localizes in the mitochondrial outer membrane (MOM) and binds with the voltage-dependent anion channel (VDAC)
[93][14].
RWe
searchers were the first to identify the role of hepatic HKDC1 in glucose metabolism. Using a mouse model of HKDC1,
researcherswe demonstrated that hepatic HKDC1 modulates glucose metabolism and insulin sensitivity in mice. Although HKDC1 has nominal expression in normal hepatocytes
[94][17], it is significantly upregulated in hepatocellular carcinoma (HCC) cells
[95][96][149,150], implying that it plays an essential role in HCC. By using HKDC1 knockout models,
rwe
searchers have shown that cellular HK activity is not affected by HKDC1 ablation; however, there is a significant increase in glucose uptake, where the bulk of glucose carbons flow through the glycolytic shunt pathways PPP and HBP (
Figure 2)
[33][16].
ResWe
archers further show that HKDC1 interacts with the mitochondria, and its loss results in mitochondrial dysfunction
[33][16]. Since cancer cells require ATP to prepare for cell division during the synthetic (S) phase of the cell cycle, a deficiency in ATP may cause cell cycle arrest. Others have shown that HKDC1 is also significantly increased in breast cancer cells, enhancing glucose uptake and mitochondrial membrane potential to encourage cell survival and growth. In agreement with this phenomenon, HKDC1 knockdown increased the production of reactive oxygen species (ROS), the activation of caspase 3, and apoptosis
[97][52]. Li et al.
[98][151] used RNA-seq data from The Cancer Genome Atlas to pinpoint genetically altered genes in a univariate survival analysis of patients with squamous cell lung carcinoma (SQCLC). Seven thousand two hundred twenty-two genetically modified genes were discovered by the analysis of RNA-seq data from 550 SQCLC patients, and HKDC1 was one of 14 feature genes with more than 100 frequencies linked to a worse prognosis
[98][99][151,152]. HKDC1 mRNA and protein levels were also expressed higher in lung cancer cell lines than in healthy lung epithelial cells.
Figure 2. Schematic representation of the effects of HKDC1 over-expression in cancer cells. The cell membrane glucose transporters (GLUT 1/3) mediate the glucose uptake, which is degraded to pyruvate by glycolysis. Upregulation of HKDC1 (and other HKs) in many cancer types leads to enhanced generation of glycolytic intermediate, which functions as precursors for numerous metabolic pathways necessary for the biosynthesis of cellular components; pentose phosphate pathway (marked with thick red arrows), cholesterol biosynthesis, and fatty acid biosynthesis. Notably, HKDC1 upregulation leads to an increase in HKDC1-mitochondrial binding, which is responsible for the maintenance of glycolysis and TCA cycle and contributes to unabated cell proliferation through the aversion of apoptosis and endoplasmic reticulum (ER)-mediated stress response mechanisms by reducing the number of physical contact points between ER and mitochondria.
Additionally, there was a direct correlation between the degree of HKDC1 protein expression and histological differentiation, reduced survival, tumor size, pN (N refers to the number of nearby lymph nodes with cancer) stage, and poor prognosis. In agreement with these results, lung cancer cell lines stably overexpressing HKDC1 demonstrated increased glucose consumption, lactate generation, proliferation, migration, and invasion compared to healthy lung epithelial cells
[98][99][151,152]. A comparison study on RNA sequencing (RNA-Seq) analysis of colorectal cancer (CRC) and matched standard tissue samples has observed significant splicing variations in nine genes in CRC. Interestingly, the authors discovered alternate regulation of the first exon in HKDC1 using exon sequencing (DEXSeq) to uncover variations in relative exon usage. HKDC1 E1a-E3a was elevated in CRC, suggesting a potential functional impact because of a projected change in the HKDC1 protein sequence
[100][101][102][153,154,155]. Another study has reported a 13 h phase change in HKDC1 expression between SW480 cells and their metastatic counterpart SW620 (a core clock gene) that occurs in conjunction with a phase shift in aryl hydrocarbon receptor nuclear translocator-like protein-1 (BMAL1). In SW480 cells, silencing BMAL1 results in an elevation of HKDC1 expression, and this effect was eliminated in SW620 cells. These findings imply that HKDC1 and the circadian clock interact, as the circadian clock is altered in metastatic cells
[103][156].
Eukaryotic cells adjust to cellular stress by phosphorylating eukaryotic translation initiation factor 2 alpha (eIF2), which results in the translation of specific transcripts that enable the cell to withstand stress
[66][67][68][69][104][105][123,124,125,126,157,158]. Activating transcription factor 4 (ATF4) is a leucine zipper transcription factor that modulates the cellular integrated stress response to allow cells to adapt to and endure stressors
[76][106][107][133,159,160]. The overexpression of ATF4 causes the HKDC1 gene transcription to increase significantly under cellular stress, changing hepatocyte mitochondrial dynamics
[108][161]. HKDC1 is upregulated in response to the endoplasmic reticulum (ER) stress or mitochondrial respiratory chain inhibition; however, when these stressors are present in combination with RNA interference to decrease ATF4, HKDC1 gene expression is reduced
[108][161].