The diagnostic and clinical success of standardization of BCR-ABL1 p210 monitoring in chronic myeloid leukemia patients could be seen as a good example for further standardization of molecular monitoring in other gene rearrangements. This article aims to summarize the steps in the diagnosis and molecular monitoring of p210 BCR-ABL1, as well as to consider the possible future application of a more sophisticated method such as digital polymerase chain reaction.
- BCR-ABL1
- Chronic myeloid leukemia
- MMR
- conversion factor
- international scale
- molecular monitoring.
1. Introduction
Chronic myeloid leukemia (CML) is a myeloproliferative neoplasm with an increased proliferation and accumulation of leukocytes and precursor myeloid cells in bone marrow (BM) and peripheral blood (PB) with an annual age-adjusted incidence of 1.6 per 100,000 population [1]. CML is mainly characterized by t (9; 22) (q34; q11) chromosomal translocation [2], giving rise to BCR-ABL1 p210 fusion protein with constitutive activation of tyrosine kinase activity. In most CML patients (~95%), the BCR-ABL1 rearrangement arises from two major breakpoints, involving exons 13 or 14 of BCR and exon 2 of ABL1 (e13a2 and e14a2) [2].
In the beginning, CML patients were treated with chemotherapy agents such as cytarabine, hydroxyurea, and immunomodulatory agents like interferon-alpha. Nowadays, the standard of care is tyrosine kinase inhibitors (TKIs), which considerably improved response rates, survival, and quality of life of patients [3].
Cytogenetic analysis or fluorescence in situ hybridization (FISH) is important at diagnosis and until a Complete Cytogenetic Remission (CCyR) is achieved. After the introduction of TKIs, most patients reach a deep molecular response (DMR) that far exceeds the sensitivity of a cytogenetic evaluation [4][5][6][4,5,6]. In this scenario, BCR-ABL1 mRNA levels has become the most important molecular marker for the minimal residual disease (MRD) evaluation, defining the level of molecular remission and guiding clinical decisions, such as therapy changes or discontinuation [7]. Thus, the accurate monitoring of the BCR-ABL1 fusion transcript appears mandatory to follow TKIs-treated CML patients.
The modern idea of molecular monitoring in patients with CML was introduced 20 years ago when the process of international standardization started. It is impossible to separate the standardization of molecular monitoring process and the introduction of TKI therapy in CML patients. Together, these activities have created complex pathways of inter- and intra-laboratory collaboration with the fundamental aim of optimizing and equalizing criteria for diagnosis and clinical interpretation of the results.
Herein, we retraced the steps that allowed the standardization of BCR-ABL1 monitoring from qualitative to quantitative methods.
2. Nested PCR
−3
−4
−5
−6
3. Quantitative Real-Time PCR
During the years, MRD monitoring has become increasingly important: it helps to predict treatment resistance and guide the course of treatment. It was clear the need to overcome the limits of a qualitative measurement, to move on to quantitative monitoring of BCR-ABL1 transcript with an aim to more favorable follow of the course of the disease.
In contrast with conventional PCR, in which the amplicon is detected by an end-point analysis, quantitative Real-Time PCR (qRT-PCR) allows analyzing the amplification during the exponential phase of the reaction using fluorescent molecules [11]. The cycle threshold (Ct) represents the cycle number when the fluorescence of a specific PCR product can be detected above the background signal. Ct values are inversely proportional to the amount of target nucleic acid in the sample. qRT-PCR also allows to obtain an absolute quantification: the initial amount of evaluated transcript can be calculated by comparing the Ct value with a standard curve generated with different known copies of target DNA [12][13][12,13].
With the introduction of qRT-PCR in routine practice, the BIOMED-1 program initiated a large study group with the purpose to standardize procedures for MRD monitoring [10].
3.1. Selection of Control Genes
3.1. Selection of Control Genes
One of the most important issues encountered with the introduction of qRT-PCR was the choice of a control gene (CG). A suitable CG can be defined as a gene with a stable expression, not affected by any experimental conditions, and should not show any pseudogenes [14][15][16]. In the early 2000s, the Europe Against Cancer (EAC) program started the first collaborative study aimed at selecting and validating a suitable CG to monitor BCR-ABL1 transcript by qRT-PCR [17]. In this study, all members of the network used the same sequence detection system and the same procedures; samples and plasmids were centrally prepared and distributed to the laboratories. Fourteen potential CGs were evaluated and, among these, only three were selected for a deep analysis: Abelson (ABL1), beta-2-microglobulin (B2M), and glucuronidase beta (GUSB). The EAC did not evaluate the breakpoint cluster region protein (BCR), already used and validated by several groups [18]. Beillard et al. identified the ABL1 gene as the most reliable CG because it was similarly expressed in normal and pathological samples and its correlation with BCR-ABL1 expression was the highest observed compared to B2M and GUSB. Nevertheless, a slight inaccuracy in measurement was highlighted of BCR-ABL1/ABL1 ratio at a high level of fusion transcripts, due to the co-amplification of ABL1 and BCR-ABL1 [17]. In the same year, Gabert et al. demonstrated that using a CG to normalize results greatly improved the data reproducibility between laboratories [19]. Furthermore, they established the reference ranges for the CGs in normal PB, BM, and PB stem cells [19]. Successively, Rulcova et al. [20] performed an independent analysis of the suitability of ABL1, B2M, and GUSB as CG to monitor CML patients. In contrast with Beillard et al. [17], they highlighted several perplexities about the use of ABL1, while they identify B2M as better CG [20]. Indeed, they confirmed and underlined that BCR-ABL1/ABL1 results were not linear when BCR-ABL1 levels are over 10%, probably because EAC primers also amplify BCR-ABL1. Although this criticism was already described and shared by the CML international community [17][21], it did not find a large consensus, because a precise quantification of BCR-ABL1 levels over 10% had poor clinical relevance.
In recent years, different papers highlighted that the time of declining of BCR-ABL1 transcript can result in distinctly different prognostic subgroups, prompting the implementation of dynamical parameters and the use of an alternative control gene [22][23]. Recently, Moisoiu et al. suggested the use of a mathematical transformation to mitigate this situation, continuing to use ABL1 as CG. Indeed, they propose to use a correction that considers the distinction between ratio and proportion. This correction can be applied to all the range of BCR-ABL1/ABL1, but it results as significant only when this percentage is over 10%. Using this strategy, ABL1 can be used as CG also when BCR/ABL1 IS values are > 10%, returning correct results [24].
3.2. International Standardization of p210 qRT-PCR Results
In the early 2000s, the International Randomized Study of Interferon versus STI571 (IRIS study) was conducted [18] to test imatinib versus interferon-cytarabine. After 12 months of treatment, imatinib has shown preponderance efficacy and significantly higher rates of transcript reduction in patients who had achieved complete cytogenetic remission [18]. It was also found that patients who had obtained at least 3-log reduction from the initial BCR-ABL1 transcript level had a likelihood of progression-free survival of 100% in the next 24 months [18]. Moreover, the 3-log reduction from the initial transcript level was defined as a major molecular response (MMR) [18].
During this trial, qRT-PCR, the gold standard for molecular diagnosis, was performed in three reference laboratories: Adelaide, London, and Seattle. The CG of choice in each lab was BCR and the results were expressed as a percentage of BCR-ABL1/BCR.
Due to the huge heterogeneity in the qRT-PCR steps among the three reference laboratories, it was observed a significant inter-laboratories difference in the reproducibility of both the pathological and control genes. Thus, the main problem was the interpretation of the differences between the obtained results.
To overpass this problem, the leading scientists in the IRIS study established that each reference laboratory should create its own assortment of the same 30 newly diagnosed untreated CML patients to extrapolate median value. This laboratory-specific median value was assumed as a standardized baseline, corresponding to 100% BCR-ABL1/ABL1. The 3-log reduction was defined as a reduction from the laboratory-specific median value and not as a log reduction calculated from the BCR-ABL1 level at the time of diagnosis for every newly diagnosed patient. In this way, the results of the three reference laboratories were normalized towards the standardized baseline.
A few years later, in 2005, an international consensus meeting was held in Bethesda, USA [25]. This can be seen as a milestone point associated with the expression of BCR-ABL1 measurements on an international scale (IS). Regarding this, when the values of the BCR-ABL1 transcripts produced anywhere worldwide are expressed on an IS, they could be more likely comparable. However so far, there were only two criteria on which the IS could be fastened: the definition of standardized baseline and MMR (
3.2. International Standardization of p210 qRT-PCR Results
Figure 1).
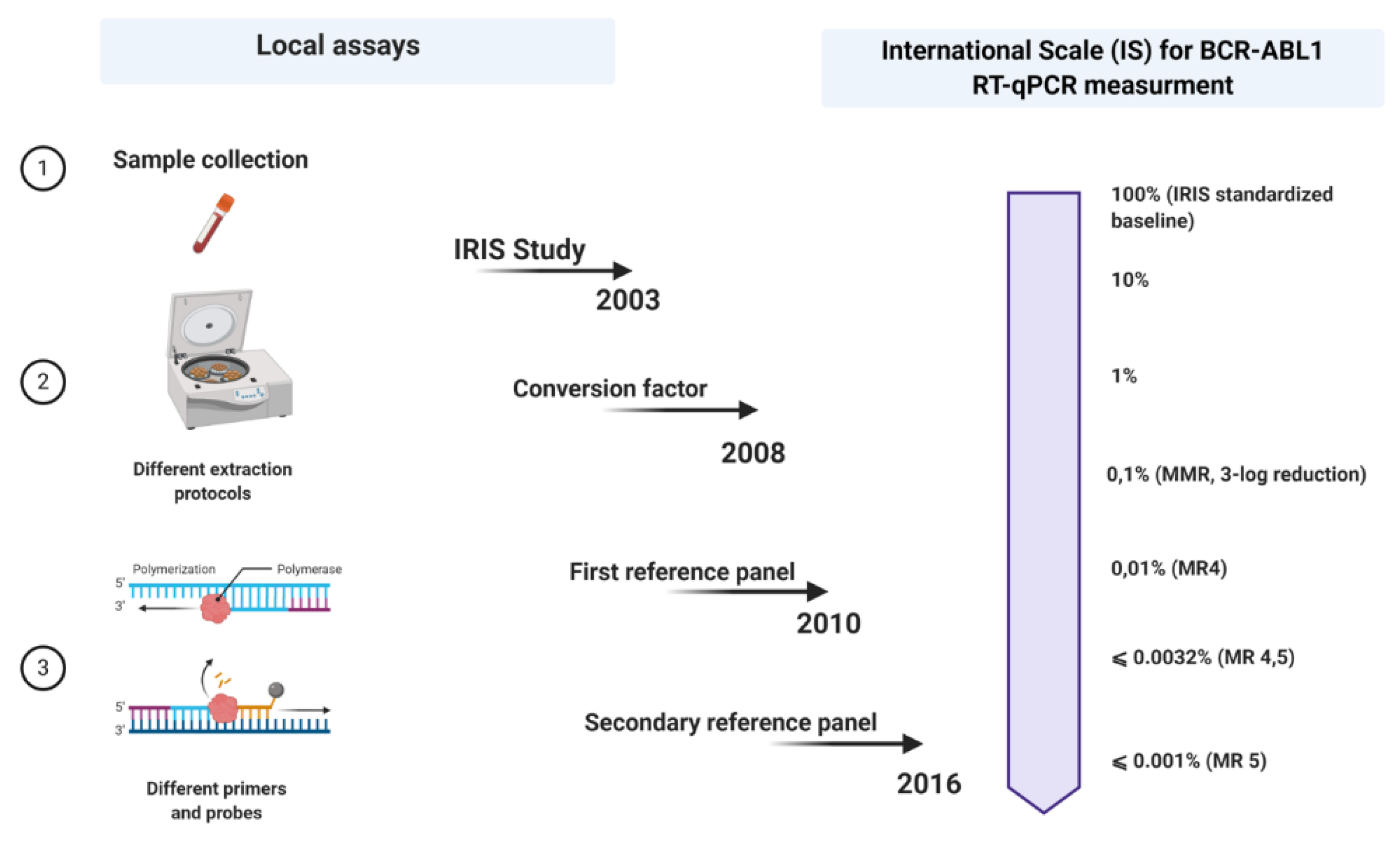
Figure 1.
Meanwhile, the 30 samples used during the IRIS study were no longer available, so a new reasonable replacement had to be found. In favor of that, two laboratories, one from Germany and the other from Australia, exchanged RNA and cDNA samples, reproducing the standardized baseline using the ABL1 as a control gene [26]. The BCR-ABL1/ABL1 ratio obtained in the German laboratory among the patients who have achieved MMR was 0.12%, corresponding with the 3-log reduction from the standardized baseline established in the IRIS trial, fixed at 0.10% (MMR) [25]. The level of 1% approximately corresponds to the achievement of CCyR [25].
3.3. The “Era” of Conversion Factor
The definition of standardized baseline and MMR paved the way for expressing the results in a standardized international numerical scale (IS), more practical than having results expressed as “log reduction”. Although the idea of an IS seemed revolutionary, its implementation proved to be a major challenge. Differences in the methods adopted by various laboratories determined great variability in the results obtained. To face this problem, the EAC group had previously started a standardization of the qRT-PCR protocol. Despite their efforts to reduce variability, significant differences between results were still present [19].
The calculation of a laboratory-specific conversion factor (CF) was intended to equalize these discrepancies. Branford et al. explained the first attempt to establish validated CFs [27]. In a big collaborative study, Adelaide was the reference laboratory, and 39 satellite laboratories participated in this project. The Bland and Altman method was used to calculate a CF for each lab, comparing the dataset from the same samples generated by the participant laboratories with that obtained by the reference laboratory (
3.3. The “Era” of Conversion Factor
3.4. Development of Reference Panels and Commercial Kits
In favor of easier access to the IS, developing a reference panel and/or standard kit for molecular diagnosis would solve the problem of CF unsustainability over time. In 2010, the World Health Organization (WHO) developed the first International Genetic Reference Panel for the quantification of BCR-ABL1 mRNA [
29]. Ten laboratories, all with pre-validated CFs, participated in this study, using the three most used control genes: ABL1, BCR, and GUSB. The creation of this reference panel is based on the preparation and titration of different dilutions of freeze-dried cell lines, such as HL-60, as BCR-ABL1 negative, and K562, as BCR-ABL1 positive. The material was prepared in 4-step dilutions (10%, 1%, 0.1%, and 0.01%) of K562 cells in the HL-60 cell line, considering that these dilutions are enough to provide more robust statistical results (regression line). About 3500 vials were produced for each level of dilution. Since the worldwide number of laboratories that diagnose patients with CML is around 500, the primary reference material is not sufficient to meet the full needs [29]. In fact, the primary reference material was reserved only to produce secondary reference reagents. Meanwhile, several secondary reference panels have been developed that allowed the expression of the results on IS ][30][31][32].
4. Digital PCR
Despite oncologists started talking about dPCR in 1999 [34], it has only recently appeared to be a suitable alternative or completion for qRT-PCR in disease monitoring, especially for low levels of disease [35][36][35,36]. dPCR works with the same primer sets, fluorescent molecules, and reagents of qRT-PCR, but it is characterized by a massive partition of the sample into single PCR reactions [37]. It also allows performing an absolute quantification without the need for standard curves or reference material. In addition, dPCR results are evaluated by an end-point analysis, reducing error rates by removing the amplification efficiency dependence of qRT-PCR [38]. Indeed, dPCR is independent by primer efficiency, in respect to qRT-PCR, where primer efficiency has a huge impact on accurate quantification. dPCR workflow is based on three main steps: massive partition of the target, end-point PCR, and Poisson statistics (Figure 2) [39]. Currently, different commercialized platforms are available, principally divided into two categories: micro-well chip-based or microfluidic-chamber-based (cdPCR) and droplet dPCR (ddPCR) technologies. These platforms differ from the method used to partition the samples: cdPCR is based on a physical subdivision, while ddPCR is based on emulsion PCR technology [38].
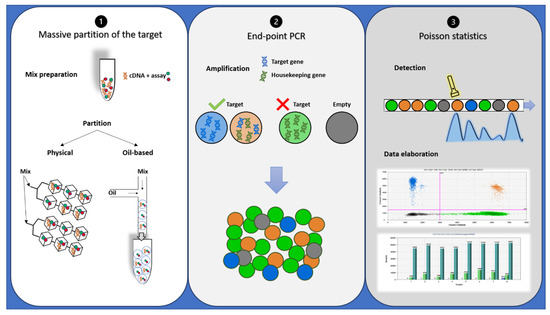
Figure 2. Description of droplet PCR workflow: (1) PCR reaction mixtures for each sample were partitioned in a microfluidic chip or through an oil-based emulsion. (2) The partitioned samples were placed into a standard thermal cycler for end-point PCR amplification: In the droplets containing target cDNA, the specific probe hydrolysis occurs and bright fluorescence appears, while in the droplets containing no target molecules (empty), only background probe fluorescence results. (3) Each droplet’s fluorescence was detected and processed into a two-dimensional scatter plot display. The number of droplets within each gate was then counted.
5. Conclusions
CML is the first human cancer that was treated with target therapy, TKIs, and in this way, the molecular monitoring of treatment efficacy has become essential.
The successful clinical use of the standardized e13a2 and e14a2 fusion transcripts in monitoring of CML patients so far could be seen as good example for further standardization of molecular monitoring in other gene rearrangements. This should be considered not only in the onco-hematology field, but also in the other oncology diseases where specific gene rearrangements are presented.