Genetics play a substantial role in epilepsy. This study presents a panoramic view of genetics known so far that contributes to common epilepsies and how the use of genetics has evolved in clinical implications of the field for disease prognosis, diagnosis as well as in treatment management. Discussing the risk factors – both genetic and non- genetic, or a combination of both, and how it can be a vital tool for prognosis or diagnosis of different endo-phenotypes of common epilepsies. Such markers hold the potential for genetic diagnosis. So, here we discussed how far are we from achieving this goal of precision medicine and clinical utility in real-time settings.
- common epilepsies
- epilepsy
- seizures
- genetics
- genetic biomarker
- diagnosis
- prognosis
- pharmacogenomics
- precision treatment
- molecular markers
1. Introduction
Epilepsy, one of the common neurological disease, is characterized by recurrent unprovoked seizures, affecting people of all age, gender, race and geographical location. Nearly 50 million people are affected worldwide with a prevalence rate of 5–10 per 1000 people [1] which accounts for more than 0.5% of the global burden of the disease [2]. Due to the vast heterogeneity associated with the genetics of common epilepsies, it is diagnosed by clinical phenotyping. There are other risk factors like age at onset, and comorbidities (such as psychiatric disorder, intellectual disability and others) that contribute to epilepsy etiology. With time, the disease classification has been dynamic, in coherent with the updated research findings. Based on seizures, epilepsy classification has evolved into different types which are: focal epilepsy, generalized epilepsy, combined focal and generalized epilepsy, and unknown epilepsy. Additionally, according to etiology it is classified as structural, genetic, infectious, metabolic, immune and unknown [3].
Traditional genetic studies like candidate gene study, linkage study, genome-wide association studies (GWAS) are based on screening of the whole genome for the genotype—phenotype association to get insight about genetic architecture and disease susceptibility of complex disease. Several GWAS have been performed to explore the disease genetics in epilepsy. Initially, the findings of GWAS were largely negative due to the small sample size and high genetic heterogeneity in epilepsy [4],[5]. Later, several international consortiums joined hands for integrative efforts to explore the disease genetics and its implications. These collaborative efforts of different groups working in the international epilepsy research community made two major advancements in the field. First, large sample size in studies assured a sufficient statistical power for successful genetic association. One of the prominent consortiums established to address the inconsistencies in genetic data is the ILAE. This group published the largest GWAS to date that included 15,212 epilepsy cases and 29,677 controls resulting in the identification of 16 statistically significant risk alleles [6]. Secondly, the consortiums provided a broader phenotypic spectrum by utilizing multi-disciplinary approaches to understand the disease heterogeneity. With this iterative process of cooperative effort, ILAE came up with the newer classifications of epilepsy based on seizure type, disease etiology and clinical factors [7] after almost three decades since the last ILAE classification in 1989 [8]. Several other consortiums have laid the foundation in this field, for example Epi4K majorly explores the genetics of common forms of epilepsy and epileptic encephalopathies, including the prognostic determinants of these disease. Another consortium, Epi25K is the unification of Epi4K, EPIGEN, EuroEPINOMICS, the Epilepsy Phenome/Genome Project, EpiPGX, SANAD, and EpiCURE consortiums which aims to combine the genotype, phenotype, and genomic sequencing data and to perform joint analyses of the data to expedite genetic biomarker discovery in all epilepsies.
2. Clinical Implication and Relevance of Genetic Findings
Remarkable advancements in identifying the causative mutations and technological growth in epilepsy genetics have channelized genetic testing into clinics. Various systemic level biomarker can be used to ensure proper diagnosis of the disease and the prediction of the treatment outcome (prognosis of anti-epileptic drug response) (Figure 31). Genetic biomarker is used to predict the risk occurrence of epilepsy in a person with a family history of epilepsy which is a commonly encountered situation. This can be used for pharmaco-response for selected antiepileptic drugs. Performing testing of specific single gene for diagnostics is no longer a practical approach for complex diseases like common epilepsies. Thus, development of gene panels and beginning of NGS-based or exome sequencing platforms for disease diagnosis has marked its way for a more comprehensive assessment of the disease status. These platforms predict the putative pathogenic variants of genes having a role in specific epilepsy subtype, and also of those genes with no known evidences with disease, but might regulate other genes with known functions in epilepsy. This may help us investigate the potential pathogenicity of such variants. Such studies hint that along with genetic data, clinical phenotyping, family history/ genealogy data together can assist in precision medicine in epilepsy. Clinical phenotyping does not include only neurological examination and tests but also thorough assessment of seizure type, duration and frequency, dysmorphic features, cutaneous signs, congenital malformation, variable symptoms of any other organ or organ system impairment, results of radiological, biochemical and other testing, cognitive functioning information etc. These phenotypes are influenced by several genes, epigenetic and environmental factors. Hence, gene/variant interpretation are crucially dependent on the full phenotypic picture of the patient. Therefore, these genetic variants need to be analyzed and integrated with detailed clinical phenotyping to make a diagnosis. These changes will make improvements in diagnosis and treatment.

Figure 31. Potential biomarkers for epilepsy diagnosis. Biological levels and sources of epilepsy biomarker omics can be measured across different biological levels around the genome, pharmacogenome, transcriptome, proteome and metabolome. Other than these hormones, cytokines and electrical imaging records can also act as biomarkers for disease prediction, drug response improvement and avoidance of adverse side effects of drugs. Abbreviations: miRNA: microRNA, CYP2C9: cytochrome P450 family 2 subfamily C member 9, HLA-B: major histocompatibility complex class I, B, HMGB1: high mobility group box 1, HSP70: heat shock protein 70, S100ßP: S100 calcium-binding protein B, MMP: matrix metallopeptidase 2, NSE: neuron-specific enolase, IL-1β: interleukin-1 beta, IL-1Ra: interleukin-1 receptor antagonist, HFO: high-frequency oscillations, MRI: magnetic resonance imaging, FA: fractional anisotropy, DTI: diffusion tensor imaging, fMRI: functional magnetic resonance imaging, PET: positron emission tomography, SPECT: single photon emission computed tomography. References: a[9]; b[10]; c[11]; d[12]; e[13]; f[14]; g[15]; h[16]; i[17]; j[18]; k[19]; l[20]; m[21]; n[22]; 0[23]; p[24]
Potential biomarkers for epilepsy diagnosis. Biological levels and sources of epilepsy biomarker omics can be measured across different biological levels around the genome, pharmacogenome, transcriptome, proteome and metabolome. Other than these hormones, cytokines and electrical imaging records can also act as biomarkers for disease prediction, drug response improvement and avoidance of adverse side effects of drugs. Abbreviations: miRNA: microRNA, CYP2C9: cytochrome P450 family 2 subfamily C member 9, HLA-B: major histocompatibility complex class I, B, HMGB1: high mobility group box 1, HSP70: heat shock protein 70, S100ßP: S100 calcium-binding protein B, MMP: matrix metallopeptidase 2, NSE: neuron-specific enolase, IL-1β: interleukin-1 beta, IL-1Ra: interleukin-1 receptor antagonist, HFO: high-frequency oscillations, MRI: magnetic resonance imaging, FA: fractional anisotropy, DTI: diffusion tensor imaging, fMRI: functional magnetic resonance imaging, PET: positron emission tomography, SPECT: single photon emission computed tomography. References: [1][9]; [2][10]; [3][11]; [4][12]; [5][13]; [6][14]; [7][15]; [8][16]; [9][17]; [10][18]; [11][19]; [12][20]; [13][21]; [14][22]; [15][23]; [16][24]
Beyond efficient prognosis or diagnosis of the disease, one of the other prime intent behind performing genetic studies is to identify novel evidence-based drug targets for future drug development. It may also allow better designing of clinical trials to standardize drug dosing, treatment outcome evaluation or toxicity profiling with respect to specific phenotypic spectrum. Given the genetic complexity of epilepsy, different genes cause specific epilepsy subtypes that are clinically indistinguishable and on the other hand, monogenic SNPs like in SCN1A cause varied phenotypes, from febrile seizure to epileptic encephalopathies. Therefore, this is the right time to proceed towards precision medicine in epilepsy. Apart from reducing the seizure frequency alone, a panoramic view of the disease mechanism with other factors like effect of common and rare variants, CNVs, polygenic risk evidences, pathways network, pharmacogenomics, other clinical phenotypes like neuro-images, EEG patterns, facilitate headway to precision medicine.
2.1. Prognosis
Variations in epilepsy genes generate variability in seizure type, epilepsy type, severities and other comorbidities. These variabilities may be due to single gene or a genetic variant (Figure 4). Genotyping along with deep phenotyping is essential in this genomic era. Genetic marker- specific prognosis is explored for rare epilepsy syndrome and is expanding for common epilepsies. It can render the diagnosis more certain in an early stage of the disease. For e.g. variations in KCNQ2 and KCNQ3 having 85% penetrance are identified as causal factor of benign familial neonatal seizures [258,259]. Genetic variants in such genes decrease the threshold membrane depolarization and increase neuronal burst [260]. Therefore, identification of variants in these genes with clinical phenotyping specific to benign familial neonatal seizures have a good prognosis, and can aid patient to get rid of seizures and medications at an early stage. But genetic variants in same genes cannot act as good biomarker for prognosis in all epileptic encephalopathy patient. In benign familial neonatal–infantile seizures patients, dominant point mutation is found in SCN2A gene, encode the alpha-subunit of the voltage-gated sodium channel NaV1.2 [261]. This point mutation results in increased neuronal excitability by gain of function [262,263]. Non-sense mutation found in the same gene result in more severe epilepsy and/or epilepsy encephalopathy, leading to a bad outcome. Thus, genetics can assist in outcome prediction in the benign familial epilepsies of childhood.
Variations in epilepsy genes generate variability in seizure type, epilepsy type, severities and other comorbidities. These variabilities may be due to single gene or a genetic variant (Figure 2). Genotyping along with deep phenotyping is essential in this genomic era. Genetic marker- specific prognosis is explored for rare epilepsy syndrome and is expanding for common epilepsies. It can render the diagnosis more certain in an early stage of the disease. For e.g. variations in KCNQ2 and KCNQ3 having 85% penetrance are identified as causal factor of benign familial neonatal seizures [25][26]. Genetic variants in such genes decrease the threshold membrane depolarization and increase neuronal burst [27]. Therefore, identification of variants in these genes with clinical phenotyping specific to benign familial neonatal seizures have a good prognosis, and can aid patient to get rid of seizures and medications at an early stage. But genetic variants in same genes cannot act as good biomarker for prognosis in all epileptic encephalopathy patient. In benign familial neonatal–infantile seizures patients, dominant point mutation is found in SCN2A gene, encode the alpha-subunit of the voltage-gated sodium channel NaV1.2 [28]. This point mutation results in increased neuronal excitability by gain of function [29][30]. Non-sense mutation found in the same gene result in more severe epilepsy and/or epilepsy encephalopathy, leading to a bad outcome. Thus, genetics can assist in outcome prediction in the benign familial epilepsies of childhood.
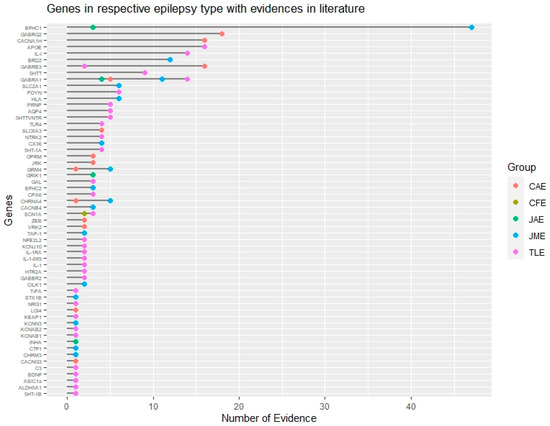
Figure 42.
Genomic landscape of common epilepsy subtypes based on evidence. Genes associated with different subtypes of common epilepsies based on their literature evidence are represented by different color codes. This figure also shows common and non-overlapping genes associated with different epilepsy subtypes. CAE: Childhood absence epilepsy, CFE: Cryptogenic focal epilepsy, JAE: Juvenile absence epilepsy, JME: Juvenile myoclonic epilepsy, TLE: Temporal lobe epilepsy.
2.2. Diagnosis
Genetic insights into the disease have given a new direction to epilepsy diagnoses directly affecting clinical care. It not only controls seizure frequency but also improves neurodevelopmental comorbidities associated with the disease. It has the prospect of wider dispersal once new targeted treatments continue to emerge based on genetic evidences. Precision diagnostics is not new in the clinical management of epilepsies which follow Mendelian inheritance but progress to genetic analysis in common epilepsies is impeded by complex pattern of inheritance. However, limited findings of candidate gene studies and GWAS have suggested role of common variants in epilepsy, nonetheless all these are causal genes/variants necessarily be susceptible to disease risk. A genetic diagnosis is very important for disease management as it avoids the unnecessary repeated blood tests, invasive biopsies, MRIs, pre-surgical workup, and even unnecessary implementation of intracranial electrodes for monitoring electrical activity of seizure. Genetic diagnosis along with the family history is very helpful in estimating the epilepsy risk for other family members that are to be tested [264]. For example, studies have shown that polymorphisms in CACNA1H gene [25,265] GABAA receptor gene with variation γ2(R43Q, rs121909673) [39], β3(P11S; rs25409), β3(S15F; rs121913126), β3(G32R; rs71651682) [46], α1(S326fs328X) [95], GABRG2(IVS6 + 2T→G) [96], GABRB3 haplotype 2 are associated with CAE and these genes are commercially used for CAE genetic testing. Commercially available genetic testing is focused on gene panels that comprises individual gene, group of genes or chromosomal loci diagnosis a specific epileptic trait. Such tests exploit the advanced NGS platforms like whole genome sequencing, NGS or targeted sequencing or others. Targeted sequencing is used for identifying genetic variants in individual gene when specific epilepsy is suspected. Epilepsy gene panel involves the analysis of group of most common genes associated with discrete epilepsy sub-types. Advantage of using gene panel is that it covers all possible genetic cause of epilepsy. Chromosome microarray involves analysis of chromosome to check any imbalance that may cause epilepsy. Endo-phenotype markers along with genetics could be useful to dissect disease complexity. Imaging endo-phenotypes and genetics provide a link between brain features and underlying genetic architecture to facilitate identification of disease related genetic variants. For e.g., photo-peroxisomal EEG response is a common observation in JME and can be a useful endo-phenotype in epilepsy gene mapping. Candidate gene studies showed association of photo-peroxisomal EEG response with BRD2 gene in JME share some neurological pathways [266]. Motor system hyper-activation and impairment of memory is observed in JME patients and their siblings, implicating trait heritability and a JME endo-phenotype [267]. Alterations of temporal cortical surface area, absence of shared thickness abnormalities and varying patterns of hippocampal atrophy is detected in family studies of TLE patients [268]. Nowadays, machine learning through neuroimaging data (resting state functional MRI, diffusion tensor imaging is being used to find specific patterns in epilepsy, enabling seizure prediction, and to distinguish between active epilepsy patient and seizure free patient. Further, using clinical data, machine learning can also predict medical and surgical outcomes through using clinical data. One assessment of a support vector machine classifier revealed a peak diagnostic sensitivity of 82.5% and a specificity of 85% by evaluating the asymmetry of functional connectivity in homologous brain regions on resting-state functional MRI in 100 patients with epilepsy and 80 controls. These endo-phenotype studies will compliment for disease category phenotype studies [269,270]. There are number of genetic tests for rare epilepsies compared to common epilepsies. Although making genetic diagnostics available to every patient is still critical and challenging. Several attempts made so far in the field of epilepsy genetic could be potential candidates that requires rigorous testing to prove safety and efficacy and demands appropriate functional validation before it is used for assessment in the clinic.
Genetic insights into the disease have given a new direction to epilepsy diagnoses directly affecting clinical care. It not only controls seizure frequency but also improves neurodevelopmental comorbidities associated with the disease. It has the prospect of wider dispersal once new targeted treatments continue to emerge based on genetic evidences. Precision diagnostics is not new in the clinical management of epilepsies which follow Mendelian inheritance but progress to genetic analysis in common epilepsies is impeded by complex pattern of inheritance. However, limited findings of candidate gene studies and GWAS have suggested role of common variants in epilepsy, nonetheless all these are causal genes/variants necessarily be susceptible to disease risk. A genetic diagnosis is very important for disease management as it avoids the unnecessary repeated blood tests, invasive biopsies, MRIs, pre-surgical workup, and even unnecessary implementation of intracranial electrodes for monitoring electrical activity of seizure. Genetic diagnosis along with the family history is very helpful in estimating the epilepsy risk for other family members that are to be tested [31]. For example, studies have shown that polymorphisms in CACNA1H gene [32][33] GABAA receptor gene with variation γ2(R43Q, rs121909673) [34] , β3(P11S; rs25409), β3(S15F; rs121913126), β3(G32R; rs71651682) [35], α1(S326fs328X) [36], GABRG2(IVS6 + 2T→G) [37], GABRB3 haplotype 2 are associated with CAE and these genes are commercially used for CAE genetic testing. Commercially available genetic testing is focused on gene panels that comprises individual gene, group of genes or chromosomal loci diagnosis a specific epileptic trait. Such tests exploit the advanced NGS platforms like whole genome sequencing, NGS or targeted sequencing or others. Targeted sequencing is used for identifying genetic variants in individual gene when specific epilepsy is suspected. Epilepsy gene panel involves the analysis of group of most common genes associated with discrete epilepsy sub-types. Advantage of using gene panel is that it covers all possible genetic cause of epilepsy. Chromosome microarray involves analysis of chromosome to check any imbalance that may cause epilepsy. Endo-phenotype markers along with genetics could be useful to dissect disease complexity. Imaging endo-phenotypes and genetics provide a link between brain features and underlying genetic architecture to facilitate identification of disease related genetic variants. For e.g., photo-peroxisomal EEG response is a common observation in JME and can be a useful endo-phenotype in epilepsy gene mapping. Candidate gene studies showed association of photo-peroxisomal EEG response with BRD2 gene in JME share some neurological pathways [38]. Motor system hyper-activation and impairment of memory is observed in JME patients and their siblings, implicating trait heritability and a JME endo-phenotype [39]. Alterations of temporal cortical surface area, absence of shared thickness abnormalities and varying patterns of hippocampal atrophy is detected in family studies of TLE patients [40]. Nowadays, machine learning through neuroimaging data (resting state functional MRI, diffusion tensor imaging is being used to find specific patterns in epilepsy, enabling seizure prediction, and to distinguish between active epilepsy patient and seizure free patient. Further, using clinical data, machine learning can also predict medical and surgical outcomes through using clinical data. One assessment of a support vector machine classifier revealed a peak diagnostic sensitivity of 82.5% and a specificity of 85% by evaluating the asymmetry of functional connectivity in homologous brain regions on resting-state functional MRI in 100 patients with epilepsy and 80 controls. These endo-phenotype studies will compliment for disease category phenotype studies [41][42]. There are number of genetic tests for rare epilepsies compared to common epilepsies. Although making genetic diagnostics available to every patient is still critical and challenging. Several attempts made so far in the field of epilepsy genetic could be potential candidates that requires rigorous testing to prove safety and efficacy and demands appropriate functional validation before it is used for assessment in the clinic.
2.3. Pharmacogenomics
The aim of pharmacogenomics is to predict how different individuals respond when prescribed with the same drug (and its dose). This aids in better clinical management of epilepsy by reducing adverse drug response and improving drug efficacy. Based on several such evidences, the USA food and drug administration (FDA) has approved drug labelling for patients with certain genetic variants. Most of them are addressed to reduce life-threatening adverse drug response. For e.g., the carriers of the rare POLG1 nucleotide substitution (p.Q1236H) may develop fatal hepatic failure when treated with sodium valproate [43]. A prospective genetic testing of such carrier patients may help clinicians identify individuals at high risk of this fatal drug toxicity. In other cases, one of the most widely studied HLA allele variant (HLA-B*15:02) predispose patients to a severe skin hypersensitivity (Stevens-Johnson syndrome/ toxic epidermal necrolysis) reaction when treated with carbamazepine [44]. According to an alert from US FDA, Carbamazepine should be avoided if patients carry at least one copy of HLA-B*15:02 allele and if patients are having carbamazepine for few months and do not show any cutaneous reaction then such patients are at low risk of developing such reaction ever from carbamazepine. Hence, alternate drugs are recommended for patients with HLA-B*15:02 or HLA-A*3101 allele[45].
A few studies also witnessed that genetic variants present in the drug metabolizing genes like CYP2C9, CYP2C19, drug transporters (ABCB1); or drug target gene SCN1A affect the binding of the drug to the receptor, and are responsible for altered drug efficacy. Genetic polymorphism in CYP2C9 with variant allele *2 and *3 reduce phenytoin metabolism by 25–50% [46]. This results in increased susceptibility of those patients to phenytoin toxicity at usual administered doses. The Clinical Pharmacogenetics Implementation Consortium (CPIC) and FDA proposed that phenytoin should not be used for patients with CYP2C9 *2 and *3 variants and HLA-B*15:02 genotype [47]. Therefore, such studies help in improving the drug efficacy, alternate AED therapy administration or dosage change providing new therapeutic strategies in future. Such findings strongly establish the role of pharmacogenomics in better epilepsy treatment management and implementation in clinical settings for specific population.
3. Conclusions and Future Direction
This is a new era for epilepsy genetics. Researchers and clinicians are joining hands, who are now swiftly moving towards evidence-based therapy for epilepsy management. Through the past decade, we have made remarkable progress towards gene discovery and innovation in technological and analytic determinants of this multi-faced disease. Once the gene attributing to the monogenic or polygenic cause of disease is clearly identified therapy can be targeted towards curing the defect contributed by the gene or compensate for the impaired molecular pathway caused by any variant of that gene. May be, that is why we have achieved much farther in monogenic epilepsies relative to its polygenic counterpart. From making progress towards identifying the cause of rare and severe epilepsy types to elucidating the role of molecular players deepens the understanding of patho-physiology of the disease. Anticipating the genetic variant type associated with epilepsy, its role in pathophysiology and quantitative assessment of the role of the genetic variant role in disease risk at individual and population level is a difficult challenge. It is causing a gap at translational level to implement genetics in targeted treatment. Hence, initiating curated registries of epilepsy patients, increasing number of multi-center, randomized, controlled trials are needed. Multi center collaboration like Epilepsy Genetic Initiative is providing a platform to bridge a gap between people with epilepsy, clinician and researchers for the advancement of precision medicine in epilepsy. This governs the way for epilepsy genetics into clinics.
This is a new era for epilepsy genetics. Researchers and clinicians are joining hands, who are now swiftly moving towards evidence-based therapy for epilepsy management. Through the past decade, we have made remarkable progress towards gene discovery and innovation in technological and analytic determinants of this multi-faced disease. Once the gene attributing to the monogenic or polygenic cause of disease is clearly identified therapy can be targeted towards curing the defect contributed by the gene or compensate for the impaired molecular pathway caused by any variant of that gene. May be, that is why we have achieved much farther in monogenic epilepsies relative to its polygenic counterpart. From making progress towards identifying the cause of rare and severe epilepsy types to elucidating the role of molecular players deepens the understanding of patho-physiology of the disease. Anticipating the genetic variant type associated with epilepsy, its role in pathophysiology and quantitative assessment of the role of the genetic variant role in disease risk at individual and population level is a difficult challenge. It is causing a gap at translational level to implement genetics in targeted treatment. Hence, initiating curated registries of epilepsy patients, increasing number of multi-center, randomized, controlled trials are needed. Multi center collaboration like Epilepsy Genetic Initiative is providing a platform to bridge a gap between people with epilepsy, clinician and researchers for the advancement of precision medicine in epilepsy. This governs the way for epilepsy genetics into clinics.
The future holds promise for progress in epilepsy genetic testing approaches that can be translated into improved disease diagnosis and treatment management for people with epilepsy. Large multi-national consortium and collaborative studies will generate huge data, which will be more valid and acceptable and help in making more accurate genotype–phenotype predictions. A panoramic approach is required for making advancement for precision medicine, which incorporate polygenic background and other non-genetic factors like microbiome, diet, optimal time for treatment, lifestyle like alcohol consumption and cigarette smoking which influence seizure threshold, sleep deprivation or stress should be considered which may enhance the success of the treatment [48]. A key focus is to develop a robust statistical genomic analysis approach that may consider the effect of variants in diverse population, demarcating the mutation patterns (allele frequency, its relative risk, and penetrance) contributing to the disease burden. This assures the application of genetics-based precision medicine in clinical settings.
References
- Epilepsy Factsheet . www.who.int. Retrieved 2020-11-13
- Beghi Ettore; The Epidemiology of Epilepsy. Neuroepidemiology 2019, 54, 185-191, 10.1159/000503831.
- Scheffer Ingrid E; ILAE classification of the epilepsies: position paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017, 58, 512-521, 10.1111/epi.13709.
- Kasperaviciūte Dalia; Common genetic variation and susceptibility to partial epilepsies: A genome-wide association study. Brain 2010, 133, 2136–2147, 10.1093/brain/awq130.
- Guo Youling; Two-stage genome-wide association study identifies variants in CAMSAP1L1 as susceptibility loci for epilepsy in Chinese. Hum Mol Genet 2011, 21, 1184-9, 10.1093/hmg/ddr550.
- International League Against Epilepsy Consortium on Complex Epilepsies; Genome-wide mega-analysis identifies 16 loci and highlights diverse biological mechanisms in the common epilepsies. Nat. Commun 2018, 9, 5269, 10.1038/s41467-018-07524-z.
- Scheffer Ingrid E; ILAE classification of the epilepsies: Position paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017, 58, 512-521, 10.1111/epi.13709.
- Commission on Classification and Terminology of the International League against Epilepsy; Proposal for revised classification of epilepsies and epileptic syndromes. Commission on Classification and Terminology of the International League against Epilepsy. Epilepsia 1989, 30, 389-99, 10.1111/j.1528-1157.1989.tb05316.x.
- Glauser Tracy A; Biomarkers for antiepileptic drug response. Biomark Med. 2011, 5, 635-41, 10.2217/bmm.11.75.Biervert C; A potassium channel mutation in neonatal human epilepsy. Science 1998 , 279 , 403–406, 10.1126/science.279.5349.403.
- An Ning; Elevated serum miR-106b and miR-146a inpatients with focal and generalized epilepsy. Epilepsy Res . 2016, 127, 311-316, DOI: 10.1016/j.eplepsyres.2016.09.019.Singh N A; A novel potassium channel gene, KCNQ2, is mutated in an inherited epilepsy of newborns. Nat. Genet. 1998 , 18 , 25–29.
- Wang Jun; Genome-wide circulating microRNA expression profiling indicates biomarkers for epilepsy. Scientific reports 2015, 5, 9522, 10.1038/srep09522.Maljevic Snezana; K V 7 channelopathies. Pflügers Archiv-Eur. J. Physiol 2010 , 460 , 277–288.
- Martins-Ferreira R; Circulating microRNAs as potential biomarkers for genetic generalized epilepsies: a three microRNA panel. Eur J Neurol 2020, 27, 660-666, DOI: 10.1111/ene.14129.Heron Sarah E; Sodium-channel defects in benign familial neonatal-infantile seizures. Lancet 2002 , 360 , 851–852.
- Paudel Yam Nath; HMGB1: A Common Biomarker and Potential Target for TBI, Neuroinflammation, Epilepsy, and Cognitive Dysfunction. Front Neurosci. 2018, 12, 628, DOI: 10.3389/fnins.2018.00628.Scalmani Paolo; Effects in Neocortical Neurons of Mutations of the Nav1.2 Na+ Channel causing Benign Familial Neonatal-Infantile Seizures. J. Neurosci. 2006, 26, 10100–10109.
- Chang Chiung-Chih; Clinical significance of serological biomarkers and neuropsychological performances in patients with temporal lobe epilepsy. BMC Neurol. 2012, 12, 15, 10.1186/1471-2377-12-15.Liao Yunxiang; Molecular correlates of age-dependent seizures in an inherited neonatal-infantile epilepsy. Brain 2010 , 133.
- Wang Rui; Serum matrix metalloproteinase-2: A potential biomarker for diagnosis of epilepsy. Epilepsy Res . 2016, 122, 114-9, 10.1016/j.eplepsyres.2016.02.009.Berkovic Samuel F; Genetics of Epilepsy in Clinical Practice. Epilepsy Curr. 2015 15, 192–196, 15 , 192–196.
- Wang Rui; Evaluation of serum matrix metalloproteinase-3 as a biomarker for diagnosis of epilepsy. J Neurol Sci . 2016, 367, 291-7, 10.1016/j.jns.2016.06.031.Liang J; Common polymorphisms in the CACNA1H gene associated with childhood absence epilepsy in Chinese Han population. Ann. Hum. Genet. 2006 , 71 , 325–335.
- Wyllie E; Postictal serum creatine kinase in the diagnosis of seizure disorders. Arch Neurol . 1985, 42, 123-6, 10.1001/archneur.1985.04060020033010.Lü Jian-jun; T-type calcium channel gene-CACNA1H is a susceptibility gene to childhood absence epilepsy. Zhonghua Chin. J. Pediatr. 2005 , 43 , 133–136.
- Mu Rong-Zheng; A Meta-Analysis of Neuron-Specific Enolase Levels in Cerebrospinal Fluid and Serum in Children With Epilepsy. Front Mol Neurosci. 2020, 13, 24, 10.3389/fnmol.2020.00024.Wallace R H; Mutant GABA(A) receptor gamma2-subunit in childhood absence epilepsy and febrile seizures. Nat. Genet. 2001 , 28 , 49–52.
- Aydin Suleyman; Time-dependent changes in the serum levels of prolactin, nesfatin-1 and ghrelin as a marker of epileptic attacks young male patients. Peptides 2011, 32, 1276-80, 10.1016/j.peptides.2011.04.021.Tanaka Miyabi; Hyperglycosylation and reduced GABA currents of mutated GABRB3 polypeptide in remitting childhood absence epilepsy. Am. J. Hum. Genet. 2008 , 82 , 1249–1261.
- Zhu Min; High Mobility Group Protein B1 (HMGB1) and Interleukin-1β as Prognostic Biomarkers of Epilepsy in Children. J Child Neurol 2018, 33, 909-917, 10.1177/0883073818801654.Maljevic Snezana; A mutation in the GABA(A) receptor alpha(1)-subunit is associated with absence epilepsy. Ann. Neurol. 2006 , 59 , 983–987.
- Kobylarek Dominik; Advances in the Potential Biomarkers of Epilepsy. Front Neurol . 2019, 10, 685, 10.3389/fneur.2019.00685.Kananura Colette; A splice-site mutation in GABRG2 associated with childhood absence epilepsy and febrile convulsions. Arch. Neurol. 2002 , 59 , 1137–1141.
- Zijlmans Maeike; High-frequency oscillations as a new biomarker in epilepsy. Ann Neurol . 2012, 71, 169-78, 10.1002/ana.22548.Koeleman Bobby P C; Photoparoxysmal EEG response and genetic dissection of juvenile myoclonic epilepsy. Epilepsy Behav. 2013 , 28 , S69–S71.
- Labate Angelo; White matter abnormalities differentiate severe from benign temporal lobe epilepsy. Epilepsia 2015, 56, 1109-16, 10.1111/epi.13027.Caciagli Lorenzo; Motor hyperactivation during cognitive tasks: An endophenotype of juvenile myoclonic epilepsy. Epilepsia 2020 , 61 , 1438–1452.
- Jr Jerome Engel; Epilepsy biomarkers. Epilepsia 2013, 54 Suppl 4(0 4), 61-9, 10.1111/epi.12299.Alhusaini Saud; Temporal Cortex Morphology in Mesial Temporal Lobe Epilepsy Patients and Their Asymptomatic Siblings. Cereb. Cortex 2015 , 26 , 1234–1241, 10.1093/cercor/bhu315.
- Abbasi Bardia; Machine learning applications in epilepsy. Epilepsia 2019 , 60 , 2037–2047.
- Zhang Jie; Pattern classification of large-scale functional brain networks: identification of informative neuroimaging markers for epilepsy. PLoS ONE 2012 , 7 , e36733.
- Stewart Joanna D; Polymerase γ gene POLG determines the risk of sodium valproate-induced liver toxicity. Hepatology 2010 , 52, 1791–1796.
- Ihtisham Kavish; Association of cutaneous adverse drug reactions due to antiepileptic drugs with HLA alleles in a North Indian population. Seizure 2019 , 66 , 99–103.
- Carbamazepine Response . www.ncbi.nlm.nih.gov. Retrieved 2020-11-15
- Silvado Carlos Eduardo; CYP2C9 polymorphisms in epilepsy: influence on phenytoin treatment. Pharm. Pers. Med. 2018 , 11 , 51–58.
- Caudle K E; Clinical pharmacogenetics implementation consortium guidelines for CYP2C9 and HLA-B genotypes and phenytoin dosing. Clin. Pharm. 2014 , 96, 542–548.
- Kearne Hugh; Tackling Epilepsy with High-definition Precision Medicine. JAMA Neurol. 2019 , 76, 1109.
- Lü Jian-jun; T-type calcium channel gene-CACNA1H is a susceptibility gene to childhood absence epilepsy. Zhonghua Chin. J. Pediatr. 2005 , 43 , 133–136.
- Wallace R H; Mutant GABA(A) receptor gamma2-subunit in childhood absence epilepsy and febrile seizures. Nat. Genet. 2001 , 28 , 49–52.
- Tanaka Miyabi; Hyperglycosylation and reduced GABA currents of mutated GABRB3 polypeptide in remitting childhood absence epilepsy. Am. J. Hum. Genet. 2008 , 82 , 1249–1261.
- Maljevic Snezana; A mutation in the GABA(A) receptor alpha(1)-subunit is associated with absence epilepsy. Ann. Neurol. 2006 , 59 , 983–987.
- Kananura Colette; A splice-site mutation in GABRG2 associated with childhood absence epilepsy and febrile convulsions. Arch. Neurol. 2002 , 59 , 1137–1141.
- Koeleman Bobby P C; Photoparoxysmal EEG response and genetic dissection of juvenile myoclonic epilepsy. Epilepsy Behav. 2013 , 28 , S69–S71.
- Caciagli Lorenzo; Motor hyperactivation during cognitive tasks: An endophenotype of juvenile myoclonic epilepsy. Epilepsia 2020 , 61 , 1438–1452.
- Alhusaini Saud; Temporal Cortex Morphology in Mesial Temporal Lobe Epilepsy Patients and Their Asymptomatic Siblings. Cereb. Cortex 2015 , 26 , 1234–1241, 10.1093/cercor/bhu315.
- Abbasi Bardia; Machine learning applications in epilepsy. Epilepsia 2019 , 60 , 2037–2047.
- Zhang Jie; Pattern classification of large-scale functional brain networks: identification of informative neuroimaging markers for epilepsy. PLoS ONE 2012 , 7 , e36733.
- Stewart Joanna D; Polymerase γ gene POLG determines the risk of sodium valproate-induced liver toxicity. Hepatology 2010 , 52, 1791–1796.
- Ihtisham Kavish; Association of cutaneous adverse drug reactions due to antiepileptic drugs with HLA alleles in a North Indian population. Seizure 2019 , 66 , 99–103.
- Carbamazepine Response . www.ncbi.nlm.nih.gov. Retrieved 2020-11-15
- Silvado Carlos Eduardo; CYP2C9 polymorphisms in epilepsy: influence on phenytoin treatment. Pharm. Pers. Med. 2018 , 11 , 51–58.
- Caudle K E; Clinical pharmacogenetics implementation consortium guidelines for CYP2C9 and HLA-B genotypes and phenytoin dosing. Clin. Pharm. 2014 , 96, 542–548.
- Kearne Hugh; Tackling Epilepsy with High-definition Precision Medicine. JAMA Neurol. 2019 , 76, 1109.