Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Eric Sohn and Version 2 by Camila Xu.
Telomeres are non-coding short repeat sequences (TTAGGG in vertebrates) which in combination with shelterin proteins protect the ends of linear chromosomes from degradation, recombination, and end fusions. Human telomeres range from 5–15 kb in length. Human Alternative Lengthening of Telomeres (ALT) cancers are often present as mesenchymal or epithelial origin in subsets of osteosarcomas, liposarcomas, glioblastomas, or astrocytomas.
- cancer
- review
- ALT
- telomere
1. Timeline of Early ALT Discoveries
The ALT pathway was first identified in a EST1 negative Saccharomyces. cerevisiae mutant in 1993. Most cells which lacked EST1, a gene which encodes a telomerase RNA-associated protein, lost the ability to replicate by telomerase and entered senescence and cell death. However, some survivors spontaneously developed a telomerase-independent maintenance mechanism. S. cerevisiae survivors exhibited tandem arrays consisting of both telomeric and subtelomeric DNA sequences which suggested amplification by homologous recombination between distant telomeric and subtelomeric DNA [1][34]. However, Saccharomyces pombe, Kluyveromyces lactis and Ustilago maydis survivors only extended telomeric DNA sequences [2][3][4][35,36,37]. Untransformed mouse embryonic fibroblasts (MEFs) were generated by extensive passaging of TERC knockout mutants [5][38]. In 2003, ALT neoplastic transformed mice cells were developed by Chang et al. [6][39]. Telomerase negative Caenorhabditis elegans were first proposed as a useful ALT model based on the observation of heterogeneous telomere lengths and telomeric circles [7][40]. As recently as 2015, C. elegans have been used to identify internal genomic regions which are necessary for telomere duplication [8][41]. The ALT pathway was first characterized in human cell lines in 1994 when Kim et al. identified two telomerase-negative SV40-immortalized fibroblasts (SW26 and SW13 [9][42]) using a novel telomerase activity assay [10][43]. The role of the ALT pathway in a subset of human cancer cell lines and tumors was further investigated in melanomas, osteosarcomas (including SAOS2 and U2OS), and carcinomas of the breast, ovary, lung and adrenal cortex [11][12][44,45]. Human ALT cancers exhibit unique biomarkers. In 1999, Yeager et al. identified ALT specific PML bodies (APBs) which facilitated co-localization of telomeric DNA and telomere binding proteins involved in recombination such as TRF1/2, RAD51, and RAD52. Telomerase negative cells exhibited APBs during immortalization but not wild-type or telomerase positive cells [13][25]. In 2009, Henson et al. developed a C-circle assay to detect extrachromosomal DNA in ALT cancers. C-circles were detected in the blood of ALT positive osteosarcoma patients. Since then, the C-circle assay is one of the main biomarkers of ALT [14][46].
In their original 1993 study, the basic mechanism for the ALT pathway was defined by Lundblad and Blackburn who identified two distinct ALT pathways in yeast survivors. The Rad51 dependent type I mechanism is more common in yeast. The Rad52 dependent type II mechanism results in heterogeneous telomeres which are more common in humans. The authors proposed that the Rad52 type II pathway in S. cerevisiae survivors required multiple rounds of telomere recombination [1][34]. In 1999, Teng et al. proposed that critically short telomeres lost telomere binding proteins and invaded a long telomere template strand to initiate telomere recombination [15][47]. Dunham et al. first demonstrated that ALT cell lines utilize inter-telomeric recombination. In the 2000 study, plasmid tags in telomeric DNA were copied from telomere to telomere in immortalized humans cells [16][7].
Since then, many molecular targets have been implicated in telomere recombination. Recombination proteins include the MRN complex, the SMC5/6 complex, and FANCM [17][18][19][48,49,50]. BLM and WRN helicases were found to facilitate telomere recombination [20][21][51,52]. Unlike BLM deficiency which promotes recombination at interstitial regions, loss of WRN promotes telomere specific recombination. WRN and TERC double knockout mouse mutants elevate telomere sister chromatid exchanges and activate ALT [21][52]. DNA damage response proteins involved in ALT activation include ATM, discovered in Atm-deficient mouse cells [22][53]. Additionally, loss of chromatin remodelers ATRX and DAXX are implicated in ALT activation [23][54]. The ALT recombinogenic potential is also dependent on telomere repeat-containing RNA (TERRA) transcription. In 2014, Arora et al. demonstrated that TERRA regulates the recombination activity of ALT telomeres by hybridizing with the telomeric C rich sequence. TERRA is regulated by RNaseH1, an RNA endonuclease which associates to telomeres in ALT positive cancers but not telomerase positive cells [24][55]. The ALT discoveries presented are summarized in chronological order in Figure 1.
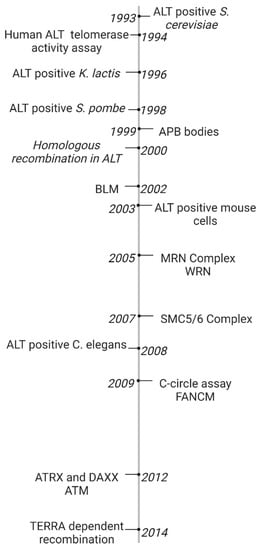
Figure 1. Timeline of ALT Discoveries. The ALT pathway was first identified in S. Cerevisiae [25][26]. A novel telomerase activity assay was used to identify the ALT pathway in human fibroblasts [26][23]. ALT was later discovered in several model organisms including K. lactis, S. pombe, mouse embryonic fibroblasts, and C. elegans [27][28][29][30][27,28,30,31]. ALT-associated PML Bodies were the first discovered hallmark of ALT cancer [13][25]. Extrachromosomal c-rich telomere circles were discovered in ALT cells using a novel C-circle assay [3][36]. One early model for the ALT mechanism hypothesized that critically short telomeres invaded a long telomere template strand to initiate telomere recombination [15][16][7,47]. ALT activation was found to be dependent on TERRA transcription [12][45]. Many proteins have been linked to telomere recombination, including BLM and WRN helicases [8][9][41,42], MRN and SMC5/6 complexes [6][39], and ATRX and DAXX chromatin remodelers [10][11][43,44], and FANCM DNA damage response protein [7][40].
2. ALT Cancer Hallmarks
ALT cancer cells can divide indefinitely (immortal cells) and exhibit break-induced repair (BIR), resulting in several biomarkers that can be used to identify ALT-positive cells. The ALT recombination mechanism results in heterogenous telomere lengths. ALT positive cell lines also exhibit high levels of extrachromosomal telomeric sequences. Circular cytosine-rich telomeric DNA (C-circles) or guanine-rich telomeric DNA (G-circles), usually <1 kb in length, correlate with ALT activity and accumulate in the nucleus [14][31][32][46,56,57]. C-circles are more than 750 times more common in ALT positive cells compared to normal and telomerase positive cells. C-circle levels are also detectable in blood samples and may be the most useful biomarker for diagnostic tests. C-circles appear to be a nonfunctional byproduct of ALT activity but a more detailed understanding of the formation of C-circles are required to contribute to a more complete model of the ALT mechanism. We speculate that DSBs produced by replication stress in telomeric DNA may create telomere fragments which self-ligate. C-circles which are 100 times more common than G-circles may result from nucleolytic degradation of the G-rich strand of T-circles. Both ALT positive and telomerase positive cancers exhibit T-circles which may be the result of T-loop fragments resolved by recombination enzymes [33][58]. The extrachromosomal DNA circles accumulate in ALT-associated PML bodies [32][57].
ALT-associated promyelocytic leukemia nuclear bodies (APBs) comprise one prominent indicator of active ALT activity [13][25]. The APB matrix is represented by a circular, hollow, membrane-less nuclear structure ranging from 50–100 nm in diameter that is formed primarily from the structural components of PML and SP100 protein [34][59]. These structures are held together through SUMO-SIM interactions, which are defined primarily as the intramolecular interactions between small ubiquitin-related modifications (SUMO) and SUMO interacting motifs (SIM) [34][59]. To this complex, telomeric DNA, related proteins, and DNA damage factors are recruited. PML depletion eliminates ALT telomere clustering and synthesis, suggesting that APBs are the location where homologous recombination (HR) occurs to maintain telomere length [35][60]. Assuming the lack of an additional mechanism to recruit the BTR (Blooms syndrome helicase, topoisomerase IIIa, and RM1/2) complex to telomere ends, APBs are essential to ALT activity [35][60]. Tethering telomeres to SUMO-SIM fusion proteins and overexpression of BLM helicase results in telomere synthesis and C-circle generation, hallmarks of ALT activity [36][61]. Loss of the replication stress response proteins FANCM, FANCD2, and SMARCAL1 increases APB formation suggesting that MMS21-mediated SUMOylation of shelterin complex proteins trigger APB formation [18][37][38][39][40][49,62,63,64,65].
It has been established that the ALT mechanism relies on recombination between telomere ends and either non-sister chromatids or extrachromosomal sequences. In a previous study, a tag on a single telomere was copied onto other chromosomes ends in ALT positive cell lines but not telomerase positive cells [16][7]. Additionally, some ALT positive cells exhibit patterns of non-canonical telomere repeats, variants of TTAGGG tandem arrays, suggesting recombination with subtelomeric or other genomic sequences, and possibly extrachromosomal telomere circles [41][66]. Therefore, ALT positive cancers exhibit increased levels of sister chromatid exchange compared to normal and telomerase positive cells.
Telomeric insertions have been observed across the genome in ALT positive cells. Some spontaneous and experimentally induced DSBs are repaired by insertion of 50–1000-bp sequences derived from distant regions of the genome [42][67]. RNA transcribed from distant regions of the genome are the primary template sequences for DNA inserted into the genome [42][67]. However, the mechanism for this mutagenic form of DSB repair remains unclear.
TERRA (Telomeric Repeat-Containing RNA) is RNA transcribed from the telomeres and hybridizes with the C-rich telomeric strand to form RNA/DNA hybrid sequences (R-loops) [24][43][55,68]. These R-loops induce recombination events between the ends of chromosomes that elongate telomeres up to >50 kb [24][31][44][55,56,69]. Inhibiting TERRA transcription alleviates ALT activity [45][70]. This suggests that TERRA is a major trigger of ALT [45][70]. Additionally, TERRA R-loops form barriers to replication suggesting ALT recombination may be triggered by replication stress [24][55].
3. Replication Stress
ALT cancers exhibit elevated levels of genomic instability and replication stress, but ALT-specific causes of telomeric replication stress are not fully understood. Aberrations in telomeric nucleoprotein structures, including heterochromatin nucleosomes, shelterin complexes, R-loops, and G-quadruplexes may contribute to ALT-specific replication stress (Figure 2) [24][46][47][48][55,71,72,73].
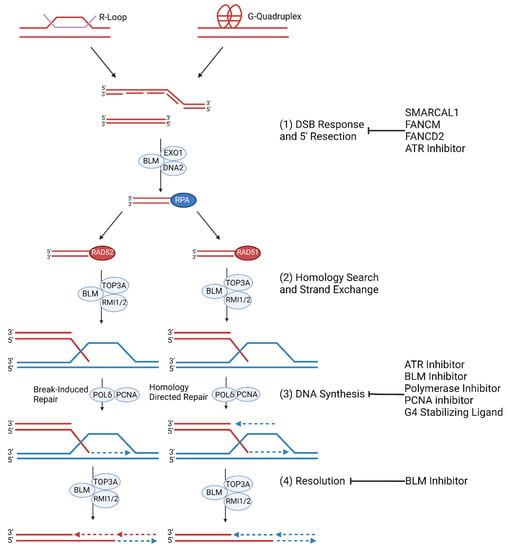
Figure 2. ALT Replication Stress and Molecular Mechanism. (1) Replication stress at telomeres is regulated by SMARCAL1 and FANCM and FANCD2. ALT cells are prone to replication stress which leads to spontaneous DNA synthesis processes. G-quadruplexes and R-loop formation at telomeres trigger replication stress which result in stalled or collapsed replication forks [49][50][74,75]. If the replication fork is not reinitiated during S/G2 phase, then mitotic DNA synthesis (MiDAS) of telomeres can occur. (2) The collapsed replication fork may be repaired by RAD52 mediated BIR or RAD51 mediated HR. (3) Telomeric MiDAS-mediated re-initiation leads to conservative DNA synthesis mediated by the BIR pathway when damaged sequences share homology with template DNA. HR- mediated re-initiation leads to semi-conservative DNA synthesis [51][76]. (4) Extended telomere ends are resolved by BLM [52][53][77,78].
Telomere heterochromatin may be regulated by a number of ALT-specific epigenetic regulators. Telomere heterochromatin decompaction appears to be a necessary but not sufficient condition for ALT activation via recombination and replication stress. ATRX (α-thalassemia/mental retardation syndrome X-linked) and its binding partner DAXX (death domain-associated protein 6) are tumor suppressing histone chaperones that promote histone H3.3 deposition and remodeling at telomeric regions. ATRX suppresses hallmarks of ALT activity such as the formation of APBs and C-circles [46][54][71,79]. Conversely, the loss of either ATRX or DAXX leads to telomeric chromatin decompaction and increased replication stress which promotes HR at the telomeres and may promote ALT activity [46][54][55][56][57][58][71,79,80,81,82,83]. ATRX and DAXX inactivation mutations highly correlate (p < 0.008 for each gene) with ALT activity in a variety of tumors including glioblastomas, oligodendrogliomas, medulloblastomas, and pancreatic neuroendocrine tumors [59][84]. Studies show that loss of ATRX also results in TERRA upregulation and G-quadruplex accumulation at telomeres [23][60][54,85]. Therefore, loss of ATRX and DAXX may be important in the initiation or maintenance of ALT replication.
ATRX regulates telomere DSB repair through two pathways involving sister chromatid cohesion or DAXX. ATRX promotes telomere cohesion via the canonical cohesion complex (SMC1-SMC3-RAD21-SA1/2) which is necessary for sister chromatid pairing during mitosis and interphase. Pairing of sister chromatids promotes DSB repair via a sister chromatid template as opposed to a homologous chromosome and prevents unequal sister chromatid recombination, interchromosomal HDR, and joining of distal ends [61][62][63][64][65][66][67][86,87,88,89,90,91,92]. ATRX deletion in mouse cells promotes defects in telomere cohesion, nonallelic telomere interactions, and homology directed repair (HDR) [68][93]. Persistent telomere cohesion during mitosis may promote T-SCEs during ALT [69][94], but the mechanism remains unclear since cohesion usually suppresses break-induced replication (BIR). ATRX also regulates telomere DSB repair through DAXX-dependent pathway. ATRX deletion in DAXX-deficient mouse cells promotes telomere damage, APB formation, and T-SCEs [68][93]. Therefore, defects in both telomere cohesion and DAXX-dependent function are necessary for telomere DSB repair associated with ALT-specific ATRX deletion [68][93]. However, some ALT cancers do not exhibit ATRX or DAXX mutations, so they are not essential to ALT cancer activation [70][71][72][73][95,96,97,98].
Loss of ATRX/DAXX may be associated with telomere insertions—fragments of tandem arrays inserted into non-telomeric regions in a subset of cancers. Some ATRX/DAXX deficient ALT-positive cancers expressed telomere insertions and telomere variant repeats. Longer telomeres correlate with higher telomere insertion event frequency [74][99]. Additionally, one study found that TERRA was transcribed from these telomere insertions [74][99]. Another possibility is that telomere insertions occur with ATRX/DAXX mutations regardless of telomere maintenance mechanisms [75][100]. Thus, the ATRX/DAXX-dependent mechanisms, rather than the ALT mechanism, may be linked to telomere insertions. However, it remains unclear whether there is a relationship between ALT cancers and telomere insertions since the methods for analyzing ATRX/DAXX mutations in ALT cancer datasets is lacking [76][101].
The nuclear receptor NR2C/F may promote ALT activity by recruiting NuRD (nucleosome remodeling and histone deacetylase) and ZNF827 (zinc finger protein 827) which deacetylate histone H3.3 and promote shelterin loss [77][78][102,103]. The unprotected telomeric strand then triggers homologous recombination (HR), a DSB repair pathway, and the remodeled telomeric strand may then promote ALT propagation [77][78][102,103]. Additionally, NR2C/F is linked to telomere insertions. NR2C/F binds to telomere variant repeats (GGGTCA) which are elevated in ALT cancers. A small subset of genomic NR2C/F binding sites can interact with telomeric repeats and serve as telomere insertion sites. These telomere fragile sites are prone to DSBs, and the unprotected ends are fused to other chromosome ends via non-homologous end joining. Fused chromosomes may be separated during mitosis, promoting more chromosome deletions, amplifications, breaks, and translocation events [79][104]. NR2C/F recruit telomeric chromatin which promotes telomere proximity that is necessary for recombination and thus ALT activity. However, NR2C/F may also drive telomere insertions by tethering NR2C/F binding sites to non-telomeric NR2C/F chromatin binding sites, resulting in recombination and insertion of telomeric variant repeats. NR2C/F accumulates in ALT-positive cells and positively correlates with increased telomeric rearrangements. Thus, NR2C/F serves to prevent telomere rearrangements and fusions by maintaining telomere integrity [80][105].
In normal cells, replication stress halts proliferation and promotes replication stress responses. FANCM (Fanconi anemia complementation group M) is a major suppressor of replication stress inducers such as R-loops. FANCM-deficient yeast cells accumulate R-loops, and ATPase inactive FANCM mutations in yeast fail to resolve R-loops, resulting in elevated levels of DSBs and ALT activity [81][82][83][18,106,107]. This suggests that FANCM interacts with FAAP24 through its ATPase domain to unwind R-loops and resolve stalled replication forks [82][106]. FANCM is also a major regulator of interstrand crosslink repair through its translocase activity. The FANCM-FAAP24-MHF1/2 complex recruits the FA core complex [84][108]. DNA lesions trigger FA to monoubiquitinate FANCD2 which localizes to BRCA1/2 and promotes HDR [84][108]. However, mutations in FANCM fail to suppress FANCD2 monoubiquitination, resulting in HDR. FANCM also interacts with PCNA (proliferating cell nuclear antigen) to remodel arrested replication forks without FA [85][86][109,110]. Similarly, SMARCAL1 (SWI/SNF-related matrix-associated actin-dependent regulator of chromatin subfamily A-like protein 1) is an ATP-dependent DNA-annealing helicase which promotes replication fork reversal and re-initiation [87][88][89][111,112,113]. Lesions and barriers in DNA hinder replication machinery, resulting in stalled replication forks. Unrepaired DNA lesions may give rise to DSBs and trigger DSB repair mechanisms and HDR-dependent ALT activity [81][90][91][92][18,114,115,116].
4. ALT Molecular Mechanism
ALT HDR is a Break-Induced Replication (BIR) pathway during the G2 and M cell cycle phases [51][93][76,117]. DSBs trigger ataxia telangiectasia mutated (ATM)-dependent checkpoint response which recruits BRCA1 complexes or 53BP1 complexes to the lesion. CTIP and MRN (MRE11-RAD50-NBS1) recruit BRCA1 to block 53BP1 from interfering with short range resection—generation of ssDNA overhangs. Exonuclease 1 executes long-range 5′−3′ resection alongside BLM (Bloom syndrome helicase) which unwinds DNA for DNA2 endonuclease. The ssDNA overhangs are filled with the ssDNA-binding factor Replication Protein A (RPA) [52][94][77,118]. Experimental BLM overexpression increases RPA at telomeres suggesting that BLM is essential for resection. RPA may then be replaced by RAD51 or RAD52 indicating a RAD51-dependent pathway and RAD52-dependent pathway. BRCA1 and BRCA2 promote recombination by mediating the exchange of RPA and RAD52 recombinase [52][77]. However, RAD52 depletion and inhibition does not affect C-circle levels suggesting an alternative RAD51 dependent pathway [95][119]. TERRA contributes to the decision to activate the RAD51 dependent pathway. A previous study showed that TERRA promotes ALT telomere synthesis at APBs via R-loops [24][55]. In RAD52 knockout conditions, TERRA maintains its ability to form R-loops. TERRA and TRF2 co-localize to APBs in both RAD52 positive and knockout conditions indicating that TERRA localization is independent of RAD52 [96][120]. Moreover, TERRA knockout or knockdown significantly reduces C-circle levels and telomere synthesis at APBs in RAD52 knockdown cells suggesting that TERRA is essential for RAD51 dependent ALT activity [96][120]. In RAD51 dependent BIR, the TERRA R-loop promotes R-loop and G-quadruplex formation which allows for the nucleoprotein overhang to replace TERRA, resulting in a transformation from an R-loop to a D-loop [96][120]. Human cancers rely on the RAD52 dependent pathway. Experimental RAD52 depletion and inhibition decreases telomeric DNA replication [51][95][76,119]. RAD51 deletion increases telomeric DNA synthesis and C-circle levels but does not affect BIR or telomere synthesis in APBs [14][95][46,119]. In RAD52 dependent BIR, the nucleoprotein 3′ overhang undergoes homology directed search and strand invasion to form a D-loop. In both pathways, the BTR complex (BLM helicase-Topoisomerase 3α-RMI1/2) is recruited to the D-loop in order to unwind the DNA. PCNA, RFC (replication factor C), and polymerase δ replisome elongates the hybridized 3′ overhang [95][97][119,121]. FANCM interacts with BTR to enable branch migration of the D-loop which may then be resolved by BTR resulting in extended telomeric DNA and a crossover event mediated by the SMX endonuclease complex (SLX1–SLX4, MUS81–EME1 and XPF–ERCC1) [52][53][81][94][18,77,78,118]. FANCM, FAAP24, or MHF1/2 depletion increases RPA, BLM, and BRCA1 localization to telomeric DNA indicating increased replication stress. siRNA-depleted FANCM also increases phosphorylated RPA and 53BP1 levels at telomeric DNA [81][90][18,114]. Co-depletion of FANCM and BLM or BRCA1 significantly decreases ALT cell viability [98][122]. BLM overexpression results in increased telomere synthesis, APBs, and C-circles, while BLM depletion results in the opposite effects [20][51]. SLX4 overexpression also results in decreased telomere synthesis, APBs, and C-circles implicating that SLX4 and BLM antagonize each other [99][100][123,124].