Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Daniel Romaus-Sanjurjo and Version 2 by Dean Liu.
Alzheimer’s disease (AD) is the most common degenerative disorder in the elderly in developed countries. GCurrently, growing evidence is pointing at endothelial dysfunction as a key player in the cognitive decline course of AD. As a main component of the blood–brain barrier (BBB), the dysfunction of endothelial cells driven by vascular risk factors associated with AD allows the passage of toxic substances to the cerebral parenchyma, producing chronic hypoperfusion that eventually causes an inflammatory and neurotoxic response.
- albumin
- Alzheimer’s disease
- cell adhesion molecules
- endothelial dysfunction
- endothelin-1
- EPCs
- metalloproteinases
- neuroimaging
- vascular alteration
- VEGFalbumin
1. Introduction
Alzheimer’s disease (AD) is the main neurodegenerative disease leading to dementia and cognitive impairment in the elderly worldwide. The classic pathophysiological hallmarks of AD are extracellular β-amyloid (Aβ) plaques and intracellular tau tangles, which eventually lead to the impairment of cognitive functions. These features progress slowly and are asymptomatic in the first stages of the disease. In fact, this fact hinders an acute premortem diagnosis if not aided by biological markers, as AD symptomatology may share similarities with other causes of dementia.
Besides cellular alterations, AD is also characterized by the presence of several vascular alterations, including small infarcts, or lacunes, due to the occlusion of branches of cerebral arteries, increases in the number of atrophic vessels and amount of vascular tortuosity (abnormal twists and turns in vessels), and decreases in microvascular density and length. Indeed, such brain vascular-associated alterations underlie many pathophysiological mechanisms of AD [1][2][1,2]. Accordingly, the two-hit vascular hypothesis points at the initial damage in cerebral vasculature (hit one) as the inducer of the accumulation of β-amyloid in the brain (hit two) [3]. Remarkably, cerebral blood–brain barrier (BBB) leakage and microbleeds are associated with cognitive decline in patients with mild cognitive impairment (MCI) and early AD, which opens the door to search for new biomarkers allowing for the diagnosis of AD before symptoms start. Importantly, many recent studies assessed the relationship between AD and stroke, another vascular-related neurological disease with a worldwide impact [4][5][4,5]. In this regard, a meta-analysis revealed that all stroke subtypes significantly increase the risk of developing AD [4]; and, more recently, that several differentially expressed genes and cellular pathways are shared by both stroke and AD [5]. Furthermore, theour group has highlighted that higher numbers of circulating endothelial progenitor cells (EPCs) within the first week following stroke have a positive impact on functional outcome [6][7][8][9][6,7,8,9], and several studies point to EPCs as a beneficial target for AD [1]. Altogether, EPCs may offer a new target to find a treatment for AD based on their recent molecular and genetic connections.
2. Vascular Alterations in AD Brains
The presence of vascular alterations entails an increased risk of developing dementia [10]. A meta-analysis of 2856 patients showed that a 60% prevalence of dementia was present in patients with macroinfarcts and lacunar disease; this percentage diminishes to 56% in small-vessel disease patients, and ranges from 57 to 70% in the presence of microinfarcts (depending on the number of lesions) [10]. More precisely, a post-mortem study found that 80% of AD patients had vascular pathology (e.g., presence of large infarcts, lacunes and multiple microinfarcts, hemorrhages, atherosclerosis, and arteriosclerosis), which has a higher prevalence than in other neurodegenerative diseases [11]. Furthermore, the presence of cerebrovascular disease, any condition affecting blood flow and blood vessel structure negatively, increases the risk of dementia in AD, with a more prominent effect in the earlier stages of the pathology [11]. Although microvascular alterations occur during normal ageing, they are especially prominent in neurodegenerative diseases, such as AD [12]. Some of these alterations include a decrease in the microvascular density, and the increase in both the number of atrophic vessels and string capillaries in AD patients [12][13][14][12,13,14]. In addition, fusiform dilatations, tortuosity, abnormal branching and fusions, as well as a reduction of the total length were detected in capillaries from AD samples [13][15][16][13,15,16]. The analysis of brain samples from AD patients has yielded a large amount of evidence indicating the presence of vascular alterations (Figure 1). For example, immunohistochemical analysis has detected the presence of potentially neurotoxic substances in the parenchyma due to their extravasation through the BBB, such as prothrombin, thrombin, fibrinogen, fibrin, albumin, and immunoglobulins [15][17][18][19][20][21][15,17,18,19,20,21]. In the specific case of prothrombin, levels of this protein in the prefrontal cortex were higher as the Braak stage increased, pointing to a greater damage of the BBB throughout the disease progression [20]. Furthermore, there is an extravasation and accumulation of immune cells in AD brains that are mediated by the overexpression of molecules such as monocyte chemoattractant protein-1 (MCP-1), intercellular adhesion molecule 1 (ICAM-1), and integrins, promoting neuroinflammation as well as BBB damage [17][22][23][24][25][26][17,22,23,24,25,26]. In addition, the release of many pro-inflammatory mediators, such as interleukin (IL)-1β (IL-1β), IL-6, IL-8, tumor necrosis factor α (TNF-α), TWEAK, and transforming growth factor β (TGF-β), into the vessels strengthen such marked neuroinflammation [17][22][27][28][17,22,27,28]. Finally, accumulation of erythrocytes and hemosiderin deposits was also detected in AD brain samples, and this is consistent with the presence of microbleeds detected by magnetic resonance imaging (MRI) [29].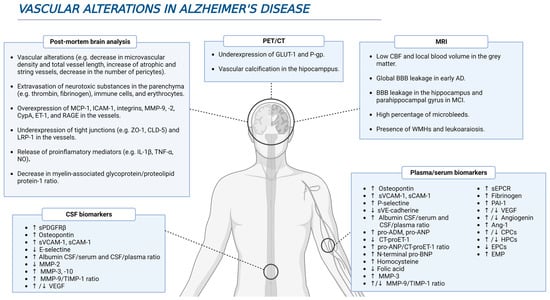
Figure 1. Several vascular-related alterations during AD. Different studies using in vivo and postmortem approaches have brought to light multiple evidence highlighting a feasible role of vascular dysfunction in AD pathophysiology.