Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Catherine Yang and Version 1 by Andrea Huwiler.
Redox-active mediators are now appreciated as powerful molecules to regulate cellular dynamics such as viability, proliferation, migration, cell contraction, and relaxation, as well as gene expression under physiological and pathophysiological conditions. These molecules include the various reactive oxygen species (ROS), and the gasotransmitters nitric oxide (NO∙), carbon monoxide (CO), and hydrogen sulfide (H2S). For each of these molecules, direct targets have been identified which transmit the signal from the cellular redox state to a cellular response. There is a cross-regulation existing between the redox mediators and sphingolipid molecules that have a fundamental impact on a cell’s fate and organ function.
- sphingolipids
- redox
- reactive oxygen species
- nitric oxide
- hydrogen sulfide
1. Nitric Oxide (NO∙)
The role of NO∙ as a signalling molecule has been extensively studied in the past [126][1]. The first direct target of NO∙ to be identified was the hemoprotein soluble guanylate cyclase (sGC) [127][2]. NO∙ binds to the heme iron of sGC forming a heme-nitrosyl complex and enzyme activation leadinr g to the formation of cGMP, which in the endothelium, is a highly potent vasodilator [128][3].
As well as binding to metal ions, NO∙ can also covalently bind to cysteines and tyrosines to form nitrosocysteine- and nitrotyrosine-modified proteins, which may cause a change in enzyme activity and function. By using the novel and sensitive methodology of mass spectrometry, multiple new proteins were identified to be modified by NO∙ causing dysfunctional signalling and contributing to diseases including cancer and inflammation [129,130,131,132][4][5][6][7].
Interestingly, when analysing pituitary adenoma tissue in a nitrosoproteome approach, the S1P lyase (SPL, Sgpl1) was identified as tyrosine nitrated on two residues, Tyr356 and Tyr366 [131][6]. Since these two sites are within the catalytic domain of Sgpl1, NO∙ modification might affect its catalytic activity, although this has not been proven yet.
Another direct target of NO∙ and a key signalling factor is the small G protein p21ras, which is cysteine nitrosated in the presence of a NO∙ donor resulting in more active p21ras and downstream signalling, such as NFκB activation [133][8] and mitogen- and stress-activated protein kinase SAPK activation [134,135,136,137][9][10][11][12].
By affecting these fundamental signalling cascades, which in turn regulate many transcription factors, NO∙ can interfere with gene transcription of many genes, including sphingolipid-regulating enzymes, and thereby alter cell responses. Indeed, NO∙ was shown to affect sphingolipid signalling in different cell types. Thus, treatment of renal mesangial cells [138][13] or endothelial cells [139][14] with NO∙ donors caused a concentration-dependent increase in cellular ceramide formation. This effect mechanistically involved upregulated activities of both aSMase and nSMase, and may occur in a similar manner as reported for ROS-activated SMase activity [94][15], i.e., directly by cysteine modification, or indirectly by GSH depletion.
In contrast to these studies, Falcone et al. [140][16] rather suggested an inhibitory effect of NO∙ on aSMase. They showed that apoptosis, induced either in dendritic cells by LPS [140][16], or in monocytic U937 cells with TNFα [141[17][18],142], involves aSMase activation and ceramide formation. This apoptotic effect of LPS and TNFα was blocked by NO∙, cGMP, and cGMP-dependent protein kinase (PKG, cGK) activation [140,141,142][16][17][18]. Such an anti-apoptotic effect of NO∙ through aSMase inhibition in tumour-associated macrophages seems also to be relevant in cancer therapy where it leads to chemoresistance [143][19]. However, the detailed mechanism of aSMase inhibition by PKG is still not clear, but at some level must involve a phosphorylation step by PKG. While many substrates of PKG have been described [144][20], aSMase has so far not been confirmed as a direct substrate of PKG. Therefore, phosphorylation of an upstream factor responsible for aSMase inhibition seems likely.
Another level of regulation of aSMase by NO∙ may exist by protein-protein interactions through nitrosated residues. In this regard, it was shown that nitrosylation of procaspase-3 promoted the direct interaction of procaspase-3 with aSMase, and this interaction had an inhibitory effect on procaspase-3 function thus inhibiting downstream caspase-8 activation and thereby reducing cell death [145][21].
Clearly, there are more factors than the SMases that determine whether cellular ceramides increase and especially the balance between SMases and CDase activities is crucial as well. Therefore, in situations when both activities of SMase and CDase increase, ceramide may not accumulate. This situation was shown for mesangial cells. When exposed to the pro-inflammatory cytokines IL-1β and TNFα, SMase and CDase activities increased in parallel thus resulting in a net unaltered ceramide [138,146][13][22]. In the case of NO∙, ceramide accumulates because SMase activities are increased while CDase activities are reduced [138,147][13][23]. This was mechanistically further approached and it seems that NO∙ causes proteasomal degradation of neutral CDase [148][24].
2. Carbon Monoxide (CO)
CO is structurally very similar to NO∙, but it is not a radical and therefore is more stable than NO∙ and diffuses freely. Endogenously, it is mainly generated as a side product of heme degradation to bilirubin by the catalytic action of heme oxygenases [149,150][25][26]. Consequently, most of the CO derives from hemoglobin and there is a constant generation of this small molecule as a result of red blood cell turnover. In addition, a small part also derives from other heme-containing proteins (hemoproteins) such as myoglobin, cytochrome c oxidase, cytochrome P450, and even nitric oxide synthases [150,151][26][27]. CO produced by nitric oxide synthases is thought to regulate neurotransmission and blood flow in the central nervous system.
In biological systems, CO reacts with reduced transition metals such as iron in hemoproteins [151][27]. High concentrations of CO are toxic and this is due to its binding to hemoglobin which occurs with many-fold higher affinity than oxygen. Consequently, CO displaces oxygen from the heme-binding resulting in impaired respiration and tissue hypoxia [151][27]. However, it is now appreciated that low concentrations of CO, as it steadily arises from red blood cell turnover, are cytoprotective. This has led to the development of CO-releasing molecules (CORM) for various therapeutic purposes [152][28] such as regulation of vascular tone, platelet aggregation, and inflammation. Mechanisms that mediate this protective effect of CO include the direct binding of CO to the various hemoproteins including sGC, cytochrome P450 proteins, cytochrome c oxidase, NADPH oxidase, and nitric oxide synthase [153][29]. Indeed, it was shown that CO and cigarette smoke, similar to NO∙, can activate the sGC yielding increased cGMP levels and endothelial relaxation [154,155,156][30][31][32].
The influence of CO on sphingolipids has only been poorly studied over the years. In one early case report of CO poisoning causing a myelinopathy, manifested by demyelination of neuronal cells, lipid analysis in the brain revealed decreased levels of total phospholipids, cerebrosides, sphingomyelins, and free cholesterol with enhanced cholesterol esters in the white matter. No changes occurred in the grey matter of the cerebral cortex [157][33]. Similarly, in a rat model of experimental acute CO poisoning, decreased brain gangliosides were detected together with changes in myelin [158][34].
Multiple stimuli can induce HO-1 expression and thereby also generate CO [153][29]. Among those, S1P was identified as an inducer of HO-1 in primary human macrophages [159][35]. This effect was mediated by the S1P1 receptor and resulted in a polarisation of macrophages to an M2 phenotype and an anti-inflammatory and anti-apoptotic reaction [159][35]. However, in other cells, such as in human leukemia cells, S1P and its acylated form, ceramide 1-phosphate (C1P), both downregulated HO-1 which was proposed to be an important mechanism in the pro-metastatic effect of these lipids on leukemia cells [160][36]. Moreover, short-chain C2-ceramide induced HO-1 in rat primary astrocytes [161][37]. This occurred through the activation of the AMPK and MAPK signalling cascades.
These few data vaguely propose that there may exist a bidirectional regulation of the sphingolipid rheostat and HO-1/CO, although a therapeutic use of this regulatory setting is not yet clear.
3. Hydrogen Disulfide
To date, only few data are available to show a direct link between H2S and sphingolipids. Interestingly, among the three H2S-producing enzymes, only CBS is converting L-cysteine to L-serine and H2S. L-serine is also the precursor in the de-novo biosynthesis of sphingolipids, and is directly used by the SPT for condensation with palmitoyl-CoA. Thus, changing the CBS expression or activity may have an impact on sphingolipid synthesis. In a recent study on isolated mouse aorta rings, it was shown that L-cysteine and L-serine have a vasorelaxant effect [162][38]. The vascular effect of both L-cysteine and L-serine was reduced by a NOS inhibitor, but also by the SPT inhibitor myriocin and the S1P1 receptor antagonist W146, suggesting the involvement of both NO∙ and S1P in the relaxant effect. It was speculated that this mechanism could be involved in the marked dysregulation of vascular tone in hyperhomocysteinemic patients (CBS deficiency) and may represent a feasible therapeutic target [162][38].
Since there is a well-studied bidirectional cross-talk between NO∙ and H2S [162[38][39][40],163,164], and in view of the cross-regulation of NO∙ on sphingolipid signalling (see Section 2.4.1), it seems very obvious that there is also a cross-talk between H2S-generating enzymes and sphingolipids.
In multiple myeloma cells, H2S donors synergistically enhanced apoptosis of cells induced by the green tea polyphenol (-)-epigallocatechin-3-O-gallate (EGCG), and thereby, potentiated the anti-cancer effect of EGCG in a mouse xenograft model [165][41]. This study further showed that in the presence of H2S donors, EGCG enhanced acid sphingomyelinase activity which was not seen by either substance alone. It was speculated that this effect on aSMase resulting in enhanced ceramide formation, is responsible for the increased apoptosis of cells [165][41].
Another interesting study showed that in human fibroblasts undergoing AKT-induced senescence, the expression of the enzyme cystathionine-β-synthase (CBS) was enhanced leading to increased H2S and GSH production, and consequently protected senescent cells from oxidative stress-induced cell death [166][42]. Especially in view of the fact that GSH is an endogenous inhibitor of nSMases, it could even be speculated that the protective effect of CBS on cell death, besides a direct protective effect of H2S, is additionally mediated by reduced ceramide formation through blocked nSMase activity.
Finally, a recent study suggested an indirect link between H2S and sphingolipids in a rat model of cerebral ischemia. In that study, the authors orally applied berberine to ischemic rats which improved the neuroinflammation and disease scores in tMCAO-induced cerebral ischemia [167][43]. It was shown that oral berberine acted on the gut microbiota and stimulated H2S production in the intestine, which subsequently activated the vagus nerve to subsequently alter the cerebral microenvironment resulting in less microglia activation and reduced neuroinflammation. Metabolomics analysis of various brain regions revealed changes in sphingolipid metabolism which may mediate the neuroprotection following vagus nerve activation. Notably, sphingosine was strongly increased in the ischemic rat cortex and downregulated by berberine treatment [167][43]. These data suggest that microbiota H2S production affects cerebral sphingolipid metabolism through vagus nerve activation. Altogether, the critical issues regarding the complex interaction of redox- and lipid-signalling described in this revisew articlearch are summarized in Figure 31 and Table 1.
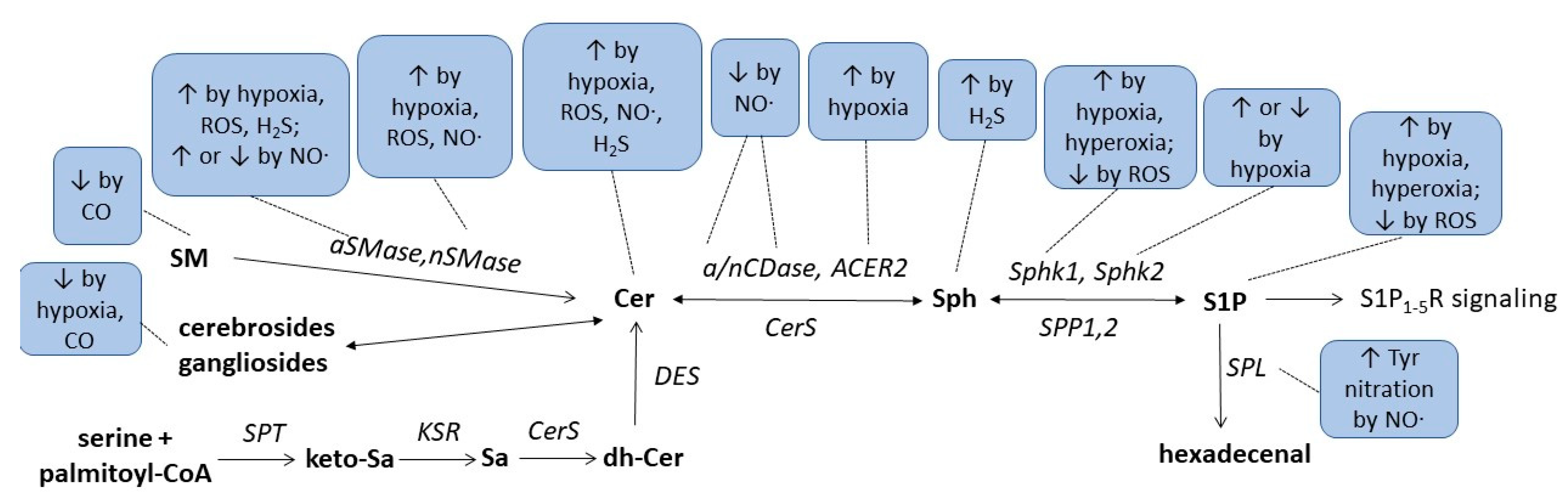
Figure 31.
Summarizing scheme of gasotransmitters, hypoxia, and hyperoxia effects on sphingolipid metabolism. For abbreviations, see text.
Table 1.
Regulation of sphingolipids and their key enzymes by redox-active mediators.
| Condition/ Enzyme |
Hypoxia | Hyperoxia | ROS | NO∙ | CO | H2S |
|---|---|---|---|---|---|---|
| Ceramides | ↑OL [52][44], CM [53][45], HC [54][46], PA [55][47] ↓VSMC [56][48] |
↑cancer cells [82,83,89][49][50][51], EC [90,91][52][53], MC [91][53], CM [53][45] |
↑MC [91,138][13][53], EC [91,139][14][53] ↓DC [140][16], U937 [141,142][17][18] |
↑cancer cells [165][41] | ||
| Sphingosine | ↑cortex [167][43] | |||||
| S1P | ↑VSMC [56][48], EC [59][54], cancer cells [58,61][55][56] | ↑mouse lung [77][57], human lung [78][58], EC [77][57] |
↓CM [121][59] | |||
| SM, Gangliosides | ↓brain [157,158][33][34] | |||||
| Cholesterolesters | ↑brain [157][33] | |||||
| Cerebrosides | ↓OL [52][44] | ↓brain [157][33] | ||||
| nSMase | ↑CM [53][45], PA [55][47] | ↑EC [90][52], CM [53][45] | ↑MC [138][13] | |||
| aSMase | ↑HC [54][46] | ↑EC [90][52] | ↑MC [138][13] ↓DC [140][16], U937 [141,142][17][18] |
↑cancer cells [165][41] | ||
| nCDase, aCDase | ↓MC [138,147][13][23] | |||||
| ACER2 | ↑adipocytes [57][60] | |||||
| Sphk1 | ↑EC [59][54], PSMC [60][61], cancer cells [58,62][55][62] | ↑mouse lung [77,79][57][63] | ↓CM [121][59] | |||
| Sphk2 | ↑cancer cells [61][56], PSMC [60][61] ↓EC [59][54] |
|||||
| SPL | Tyr nitration [131][6] | |||||
| SPT2 | ↑neuroblastoma cells [69][64] |
Abbreviations: ACER2, alkaline ceramidase 2; aSMase, acid sphingomyelinase; nCDase, neutral ceramidase; Sphk1, sphingosine kinase 1; Sphk2, sphingosine kinase 2; SPL, sphingosine 1-phosphate lyase; SPT2, serine palmitoyltransferase 2; ROS, reactive oxygen species; NO∙, nitric oxide; CO, carbon monoxide; H2S, hydrogen sulfide; EC, endothelial cells; MC, mesangial cells; SM, sphingomyelins; OL, oligodendrocytes; CM, cardiomyocytes; DC, dendritic cells; PA, pulmonary arteries; U937, pro-monocytic human myeloid leukemia cell line; VSMC vascular smooth muscle cell; PSMC, pulmonary smooth muscle cells; ↑, upregulated; ↓, downregulated.
References
- Huwiler, A.; Pfeilschifter, J. Nitric oxide signalling with a special focus on lipid-derived mediators. Biol. Chem. 2003, 384, 1379–1389.
- Arnold, W.P.; Mittal, C.K.; Katsuki, S.; Murad, F. Nitric oxide activates guanylate cyclase and increases guanosine 3′:5′-cyclic monophosphate levels in various tissue preparations. Proc. Natl. Acad. Sci. USA 1977, 74, 3203–3207.
- Gruetter, C.A.; Barry, B.K.; McNamara, D.B.; Gruetter, D.Y.; Kadowitz, P.J.; Ignarro, L. Relaxation of bovine coronary artery and activation of coronary arterial guanylate cyclase by nitric oxide, nitroprusside and a carcinogenic nitrosoamine. J. Cycl. Nucleotide Res. 1979, 5, 211–224.
- Sharma, V.; Fernando, V.; Letson, J.; Walia, Y.; Zheng, X.; Fackelman, D.; Furuta, S. S-Nitrosylation in Tumor Microenvironment. Int. J. Mol. Sci. 2021, 22, 4600.
- Zhang, Y.; Deng, Y.; Yang, X.; Xue, H.; Lang, Y. The Relationship Between Protein S-Nitrosylation and Human Diseases: A Review. Neurochem. Res. 2020, 45, 2815–2827.
- Zhan, X.; Desiderio, D.M. Nitroproteins from a human pituitary adenoma tissue discovered with a nitrotyrosine affinity column and tandem mass spectrometry. Anal. Biochem. 2006, 354, 279–289.
- Lau, B.; Fazelinia, H.; Mohanty, I.; Raimo, S.; Tenopoulou, M.; Doulias, P.T.; Ischiropoulos, H. Endogenous S-nitrosocysteine proteomic inventories identify a core of proteins in heart metabolic pathways. Redox Biol. 2021, 47, 102153.
- Lander, H.M.; Ogiste, J.S.; Pearce, S.F.; Levi, R.; Novogrodsky, A. Nitric oxide-stimulated guanine nucleotide exchange on p21ras. J. Biol. Chem. 1995, 270, 7017–7020.
- Pfeilschifter, J.; Huwiler, A. Nitric oxide stimulates stress-activated protein kinases in glomerular endothelial and mesangial cells. FEBS Lett. 1996, 396, 67–70.
- Callsen, D.; Pfeilschifter, J.; Brune, B. Rapid and delayed p42/p44 mitogen-activated protein kinase activation by nitric oxide: The role of cyclic GMP and tyrosine phosphatase inhibition. J. Immunol. 1998, 161, 4852–4858.
- Huwiler, A.; Pfeilschifter, J. Nitric oxide stimulates the stress-activated protein kinase p38 in rat renal mesangial cells. J. Exp. Biol. 1999, 202 Pt 6, 655–660.
- Lander, H.M.; Jacovina, A.T.; Davis, R.J.; Tauras, J.M. Differential activation of mitogen-activated protein kinases by nitric oxide-related species. J. Biol. Chem. 1996, 271, 19705–19709.
- Huwiler, A.; Pfeilschifter, J.; van den Bosch, H. Nitric oxide donors induce stress signaling via ceramide formation in rat renal mesangial cells. J. Biol. Chem. 1999, 274, 7190–7195.
- Huwiler, A.; Dorsch, S.; Briner, V.A.; van den Bosch, H.; Pfeilschifter, J. Nitric oxide stimulates chronic ceramide formation in glomerular endothelial cells. Biochem. Biophys. Res. Commun. 1999, 258, 60–65.
- Ratnayake, S.; Dias, I.H.; Lattman, E.; Griffiths, H.R. Stabilising cysteinyl thiol oxidation and nitrosation for proteomic analysis. J. Proteom. 2013, 92, 160–170.
- Falcone, S.; Perrotta, C.; De Palma, C.; Pisconti, A.; Sciorati, C.; Capobianco, A.; Rovere-Querini, P.; Manfredi, A.A.; Clementi, E. Activation of acid sphingomyelinase and its inhibition by the nitric oxide/cyclic guanosine 3′,5′-monophosphate pathway: Key events in Escherichia coli-elicited apoptosis of dendritic cells. J. Immunol. 2004, 173, 4452–4463.
- Barsacchi, R.; Perrotta, C.; Sestili, P.; Cantoni, O.; Moncada, S.; Clementi, E. Cyclic GMP-dependent inhibition of acid sphingomyelinase by nitric oxide: An early step in protection against apoptosis. Cell Death Differ. 2002, 9, 1248–1255.
- De Nadai, C.; Sestili, P.; Cantoni, O.; Lievremont, J.P.; Sciorati, C.; Barsacchi, R.; Moncada, S.; Meldolesi, J.; Clementi, E. Nitric oxide inhibits tumor necrosis factor-alpha-induced apoptosis by reducing the generation of ceramide. Proc. Natl. Acad. Sci. USA 2000, 97, 5480–5485.
- Perrotta, C.; Cervia, D.; Di Renzo, I.; Moscheni, C.; Bassi, M.T.; Campana, L.; Martelli, C.; Catalani, E.; Giovarelli, M.; Zecchini, S.; et al. Nitric Oxide Generated by Tumor-Associated Macrophages Is Responsible for Cancer Resistance to Cisplatin and Correlated with Syntaxin 4 and Acid Sphingomyelinase Inhibition. Front. Immunol. 2018, 9, 1186.
- Schlossmann, J.; Desch, M. cGK substrates. Handb. Exp. Pharmacol. 2009, 191, 163–193.
- Matsumoto, A.; Comatas, K.E.; Liu, L.; Stamler, J.S. Screening for nitric oxide-dependent protein-protein interactions. Science 2003, 301, 657–661.
- Franzen, R.; Pautz, A.; Brautigam, L.; Geisslinger, G.; Pfeilschifter, J.; Huwiler, A. Interleukin-1beta induces chronic activation and de novo synthesis of neutral ceramidase in renal mesangial cells. J. Biol. Chem. 2001, 276, 35382–35389.
- Franzen, R.; Fabbro, D.; Aschrafi, A.; Pfeilschifter, J.; Huwiler, A. Nitric oxide induces degradation of the neutral ceramidase in rat renal mesangial cells and is counterregulated by protein kinase C. J. Biol. Chem. 2002, 277, 46184–46190.
- Franzen, R.; Pfeilschifter, J.; Huwiler, A. Nitric oxide induces neutral ceramidase degradation by the ubiquitin/proteasome complex in renal mesangial cell cultures. FEBS Lett. 2002, 532, 441–444.
- Tenhunen, R.; Marver, H.S.; Schmid, R. The enzymatic conversion of heme to bilirubin by microsomal heme oxygenase. Proc. Natl. Acad. Sci. USA 1968, 61, 748–755.
- Ryter, S.W.; Choi, A.M. Carbon monoxide: Present and future indications for a medical gas. Korean J. Intern. Med. 2013, 28, 123–140.
- Piantadosi, C.A. Carbon monoxide, reactive oxygen signaling, and oxidative stress. Free Radic. Biol. Med. 2008, 45, 562–569.
- Adach, W.; Olas, B. Carbon monoxide and its donors—Their implications for medicine. Future Med. Chem. 2019, 11, 61–73.
- Kim, H.P.; Ryter, S.W.; Choi, A.M. CO as a cellular signaling molecule. Annu. Rev. Pharmacol. Toxicol. 2006, 46, 411–449.
- Brune, B.; Ullrich, V. Inhibition of platelet aggregation by carbon monoxide is mediated by activation of guanylate cyclase. Mol. Pharmacol. 1987, 32, 497–504.
- Arnold, W.P.; Aldred, R.; Murad, F. Cigarette smoke activates guanylate cyclase and increases guanosine 3′,5′-monophosphate in tissues. Science 1977, 198, 934–936.
- Graser, T.; Vedernikov, Y.P.; Li, D.S. Study on the mechanism of carbon monoxide induced endothelium-independent relaxation in porcine coronary artery and vein. Biomed. Biochim. Acta 1990, 49, 293–296.
- Wender, M. Studies of Cerebral Lipids in a Relapsing Case of Carbon Monoxide Poisoning. Acta Neuropathol. 1963, 3, 371–377.
- Mawatari, S. Biochemical study on rat brain in acute carbon monoxide poisoning. Folia Psychiatr. Neurol. Jpn. 1970, 24, 123–129.
- Weis, N.; Weigert, A.; von Knethen, A.; Brune, B. Heme oxygenase-1 contributes to an alternative macrophage activation profile induced by apoptotic cell supernatants. Mol. Biol. Cell 2009, 20, 1280–1288.
- Abdelbaset-Ismail, A.; Cymer, M.; Borkowska-Rzeszotek, S.; Brzezniakiewicz-Janus, K.; Rameshwar, P.; Kakar, S.S.; Ratajczak, J.; Ratajczak, M.Z. Bioactive Phospholipids Enhance Migration and Adhesion of Human Leukemic Cells by Inhibiting Heme Oxygenase 1 (HO-1) and Inducible Nitric Oxygenase Synthase (iNOS) in a p38 MAPK-Dependent Manner. Stem Cell Rev. Rep. 2019, 15, 139–154.
- Jung, J.S.; Choi, M.J.; Ko, H.M.; Kim, H.S. Short-chain C2 ceramide induces heme oxygenase-1 expression by upregulating AMPK and MAPK signaling pathways in rat primary astrocytes. Neurochem. Int. 2016, 94, 39–47.
- Mitidieri, E.; Gurgone, D.; Caiazzo, E.; Tramontano, T.; Cicala, C.; Sorrentino, R.; d’Emmanuele di Villa Bianca, R. L-cysteine/cystathionine-beta-synthase-induced relaxation in mouse aorta involves a L-serine/sphingosine-1-phosphate/NO pathway. Br. J. Pharmacol. 2020, 177, 734–744.
- Nagpure, B.V.; Bian, J.S. Interaction of Hydrogen Sulfide with Nitric Oxide in the Cardiovascular System. Oxid. Med. Cell Longev. 2016, 2016, 6904327.
- Whiteman, M.; Moore, P.K. Hydrogen sulfide and the vasculature: A novel vasculoprotective entity and regulator of nitric oxide bioavailability? J. Cell Mol. Med. 2009, 13, 488–507.
- Bae, J.; Kumazoe, M.; Yamashita, S.; Tachibana, H. Hydrogen sulphide donors selectively potentiate a green tea polyphenol EGCG-induced apoptosis of multiple myeloma cells. Sci. Rep. 2017, 7, 6665.
- Zhu, H.; Chan, K.T.; Huang, X.; Cerra, C.; Blake, S.; Trigos, A.S.; Anderson, D.; Creek, D.J.; De Souza, D.P.; Wang, X.; et al. Cystathionine-beta-synthase is essential for AKT-induced senescence and suppresses the development of gastric cancers with PI3K/AKT activation. eLife 2022, 11, e71929.
- Ni, S.J.; Yao, Z.Y.; Wei, X.; Heng, X.; Qu, S.Y.; Zhao, X.; Qi, Y.Y.; Ge, P.Y.; Xu, C.P.; Yang, N.Y.; et al. Vagus nerve stimulated by microbiota-derived hydrogen sulfide mediates the regulation of berberine on microglia in transient middle cerebral artery occlusion rats. Phytother. Res. 2022, 36, 2964–2981.
- Kendler, A.; Dawson, G. Progressive hypoxia inhibits the de novo synthesis of galactosylceramide in cultured oligodendrocytes. J. Biol. Chem. 1990, 265, 12259–12266.
- Hernandez, O.M.; Discher, D.J.; Bishopric, N.H.; Webster, K.A. Rapid activation of neutral sphingomyelinase by hypoxia-reoxygenation of cardiac myocytes. Circ. Res. 2000, 86, 198–204.
- Zhu, Q.; Lin, L.; Cheng, Q.; Xu, Q.; Zhang, J.; Tomlinson, S.; Jin, J.; Chen, X.; He, S. The role of acid sphingomyelinase and caspase 5 in hypoxia-induced HuR cleavage and subsequent apoptosis in hepatocytes. Biochim. Biophys. Acta 2012, 1821, 1453–1461.
- Cogolludo, A.; Moreno, L.; Frazziano, G.; Moral-Sanz, J.; Menendez, C.; Castaneda, J.; Gonzalez, C.; Villamor, E.; Perez-Vizcaino, F. Activation of neutral sphingomyelinase is involved in acute hypoxic pulmonary vasoconstriction. Cardiovasc. Res. 2009, 82, 296–302.
- Yun, J.K.; Kester, M. Regulatory role of sphingomyelin metabolites in hypoxia-induced vascular smooth muscle cell proliferation. Arch. Biochem. Biophys. 2002, 408, 78–86.
- Liu, B.; Hannun, Y.A. Inhibition of the neutral magnesium-dependent sphingomyelinase by glutathione. J. Biol. Chem. 1997, 272, 16281–16287.
- Mansat-de Mas, V.; Bezombes, C.; Quillet-Mary, A.; Bettaieb, A.; D’Orgeix, A.D.; Laurent, G.; Jaffrezou, J.P. Implication of radical oxygen species in ceramide generation, c-Jun N-terminal kinase activation and apoptosis induced by daunorubicin. Mol. Pharmacol. 1999, 56, 867–874.
- Gouaze, V.; Mirault, M.E.; Carpentier, S.; Salvayre, R.; Levade, T.; Andrieu-Abadie, N. Glutathione peroxidase-1 overexpression prevents ceramide production and partially inhibits apoptosis in doxorubicin-treated human breast carcinoma cells. Mol. Pharmacol. 2001, 60, 488–496.
- Huwiler, A.; Boddinghaus, B.; Pautz, A.; Dorsch, S.; Franzen, R.; Briner, V.A.; Brade, V.; Pfeilschifter, J. Superoxide potently induces ceramide formation in glomerular endothelial cells. Biochem. Biophys. Res. Commun. 2001, 284, 404–410.
- Pautz, A.; Franzen, R.; Dorsch, S.; Boddinghaus, B.; Briner, V.A.; Pfeilschifter, J.; Huwiler, A. Cross-talk between nitric oxide and superoxide determines ceramide formation and apoptosis in glomerular cells. Kidney Int. 2002, 61, 790–796.
- Schwalm, S.; Doll, F.; Romer, I.; Bubnova, S.; Pfeilschifter, J.; Huwiler, A. Sphingosine kinase-1 is a hypoxia-regulated gene that stimulates migration of human endothelial cells. Biochem. Biophys. Res. Commun. 2008, 368, 1020–1025.
- Anelli, V.; Gault, C.R.; Cheng, A.B.; Obeid, L.M. Sphingosine kinase 1 is up-regulated during hypoxia in U87MG glioma cells. J. Biol. Chem. 2008, 283, 3365–3375.
- Schnitzer, S.E.; Welgert, A.; Zhou, J.; Brune, B. Hypoxia Enhances Sphingosine Kinase 2 Activity and Provokes Sphingosine-1-Phosphate-Mediated Chemoresistance in A549 Lung Cancer Cells. Mol. Cancer Res. 2009, 7, 393–401.
- Harijith, A.; Pendyala, S.; Reddy, N.M.; Bai, T.; Usatyuk, P.V.; Berdyshev, E.; Gorshkova, I.; Huang, L.S.; Mohan, V.; Garzon, S.; et al. Sphingosine kinase 1 deficiency confers protection against hyperoxia-induced bronchopulmonary dysplasia in a murine model: Role of S1P signaling and Nox proteins. Am. J. Pathol. 2013, 183, 1169–1182.
- Ha, A.W.; Sudhadevi, T.; Ebenezer, D.L.; Fu, P.; Berdyshev, E.V.; Ackerman, S.J.; Natarajan, V.; Harijith, A. Neonatal therapy with PF543, a sphingosine kinase 1 inhibitor, ameliorates hyperoxia-induced airway remodeling in a murine model of bronchopulmonary dysplasia. Am. J. Physiol. Lung Cell. Mol. Physiol. 2020, 319, L497–L512.
- Pchejetski, D.; Kunduzova, O.; Dayon, A.; Calise, D.; Seguelas, M.H.; Leducq, N.; Seif, I.; Parini, A.; Cuvillier, O. Oxidative stress-dependent sphingosine kinase-1 inhibition mediates monoamine oxidase A-associated cardiac cell apoptosis. Circ. Res. 2007, 100, 41–49.
- Zhang, X.; Zhang, Y.; Wang, P.; Zhang, S.Y.; Dong, Y.; Zeng, G.; Yan, Y.; Sun, L.; Wu, Q.; Liu, H.; et al. Adipocyte Hypoxia-Inducible Factor 2alpha Suppresses Atherosclerosis by Promoting Adipose Ceramide Catabolism. Cell Metab. 2019, 30, 937–951.e5.
- Ahmad, M.; Long, J.S.; Pyne, N.J.; Pyne, S. The effect of hypoxia on lipid phosphate receptor and sphingosine kinase expression and mitogen-activated protein kinase signaling in human pulmonary smooth muscle cells. Prostaglandins Other Lipid Mediat. 2006, 79, 278–286.
- Ader, I.; Brizuela, L.; Bouquerel, P.; Malavaud, B.; Cuvillier, O. Sphingosine kinase 1: A new modulator of hypoxia inducible factor 1alpha during hypoxia in human cancer cells. Cancer Res. 2008, 68, 8635–8642.
- Harijith, A.; Pendyala, S.; Ebenezer, D.L.; Ha, A.W.; Fu, P.; Wang, Y.T.; Ma, K.; Toth, P.T.; Berdyshev, E.V.; Kanteti, P.; et al. Hyperoxia-induced p47phox activation and ROS generation is mediated through S1P transporter Spns2, and S1P/S1P1&2 signaling axis in lung endothelium. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 311, L337–L351.
- Kang, M.S.; Ahn, K.H.; Kim, S.K.; Jeon, H.J.; Ji, J.E.; Choi, J.M.; Jung, K.M.; Jung, S.Y.; Kim, D.K. Hypoxia-induced neuronal apoptosis is mediated by de novo synthesis of ceramide through activation of serine palmitoyltransferase. Cell. Signal. 2010, 22, 610–618.
More