Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Javier Garcia-Pardo and Version 1 by Javier Garcia-Pardo.
Parkinson’s disease, the second most common neurodegenerative disorder worldwide, is characterized by the accumulation of protein deposits in the dopaminergic neurons. These deposits are primarily composed of aggregated forms of α-Synuclein (α-Syn). PD is a complex pathology initially associated with motor deficiencies, as a result of an acute neuronal loss in substantia nigra pars compacta (SNc), with a significant dopaminergic (DA) impairment.
- Parkinson’s disease
- aggregation
- α-Synuclein
- Protein aggregation
1. Historical Overview
The history of PD began two centuries ago in England (Figure 1). In 1817, James Parkinson published a book entitled ‘An Essay on the shaking palsy’. In this treatise, Parkinson methodically described the development of a disorder in six patients with a motor disability, observing resting tremors, paralysis, and an unnatural posture [29]. Sixty years later, in 1872, the neurologist Jean-Martin Charcot, whose work helped to differentiate between bradykinesia, stiffness, and weakness in PD [30], named the disorder Parkinson’s disease in honor of James Parkinson. Two decades later, in 1893, Blocq and Marinescu analyzed a patient exhibiting resting tremors due to a granuloma that affects the SNc [31]. Finally, in 1899, Brissaud suggested that SNc is the most affected region in PD-suffering patients [32]. However, it was not until 1919 that the first pathological evidence was obtained by Trétiakoff who provided a description of significant neuromelanin loss in SNc neurons and the presence of Lewy bodies (LBs) [33], cytoplasmatic structures previously reported by Frederic Lewy in PD-affected brains [34]. Altogether, DA cell loss and LBs presence in SNc constituted the first anatomical proof of PD and allowed its post-mortem diagnosis [35].
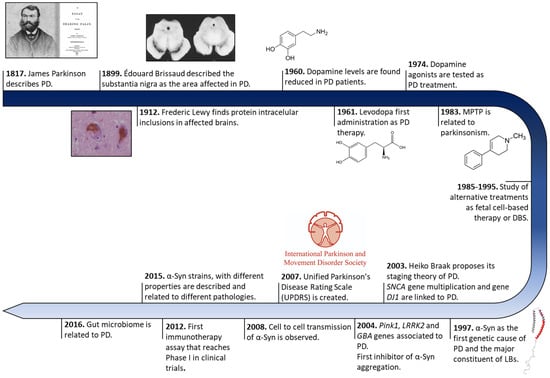
Figure 1. Historical overview of Parkinson’s disease research. Schematic representation of some of the most relevant findings in PD research from its discovery to treatment development.
Later, in 1957, Arvid Carlsson made a discovery that would play a key role in PD treatments. Carlsson described, in animals, the role of dopamine in motor activity under the control of the basal glial, whose deficiency could be reverted by L-3,4-dihydroxyphenylalanine (L-DOPA) administration [36]. Three years later, Ehringer and Hornykiewicz described a dopamine deficiency in the striatum and SNc of PD-affected brains [37]. During the following years, L-DOPA intravenous or oral administrations were extensively investigated [22], and many authors reported significant improvements in motor symptoms (MS) [23], thus it became the default therapy for PD.
Eighty years after the discovery of LBs, a protein called α-Syn was found to be the major component of these cytoplasmatic structures [21]. These findings correlated with previous evidence of genetic mutations in the SNCA gene (that encodes for α-Syn), the first gene described as a genetic cause of PD [38]. Since then, other numerous genetic alterations in SNCA (single-point mutations, duplications, and/or triplication) [39,40,41,42,43,44,45] and other genes (GBA, PINK1, LRRK2, Parkin, or DJ1, among others) [46,47,48,49] gradually appeared as risk factors of PD early onset and progression, opening up possibilities for the development of genetic models that recapitulate the molecular origin the disease better than the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) or 6-hydroxydopamine (6-OHDA) neurotoxin ones do [50]. These new cellular and animal models have allowed the study of α-Syn transmission, therapeutic approaches, and biomarkers, especially for the initial stage of PD (prodromal stage) [51,52,53,54,55]. Currently, the search for effective treatments for PD has been mainly focused on α-Syn aggregation [56], but genetic therapies and microbiota alterations linked to PD are receiving increasing attention [57,58].
2. Symptomatology
PD has been traditionally considered as a neuronal disorder with symptomatology that is limited to unilateral and asymmetric motor deficits such as rigidity, tremors, bradykinesia, and postural instability. Clinical diagnosis has been based on bradykinesia and other cardinal motor deficiencies [59,60]. The onset of these symptoms varies between <40 and >80 years of age [16]. A young onset of PD is considered to occur at an age < 45 and is usually correlated to genetic factors [61,62]. However, PD involves additional clinical features with NMS, such as sleep disorders, cognitive impairment, depression, anxiety, pain, or dementia [63,64,65,66], which significantly aggravate the patient’s quality of life [67]. The development of PD impacts numerous neurotransmission pathways, which could explain the appearance of these NMS; for example, depression could be related to the deterioration of cholinergic, noradrenergic, and serotonergic systems, while DA and noradrenergic decay would induce anxiety [68,69,70,71,72].
The absence of MS and the presence of symptoms such as olfactory dysfunction, REM sleep behavior disorder, constipation, and depression are indicators of the prodromal stage of PD [63]. The development of these NMS during the prodromal phase precedes MS by several years [63,73,74] (Figure 2). In the early stages of the disorder, patients develop bradykinesia, tremors, and rigidity, and up to 21% of them also experiment pain, depression, or anxiety [75]. Initially, the disease can be treated with symptomatic therapies, but as it progresses, the treatment becomes more complicated [61,76]. In the latest stages, patients exhibit severe NMS such as dementia (83%), hallucinosis (74%), orthostatic hypotension (48%), urinary incontinence (71%), or constipation (40%), leading to a pronounced disability [76]. In addition, these phases are characterized by a progressive physical incapacity and strong resistance to the treatments, inducing freezing gait, postural instability, falls, and choking [77].
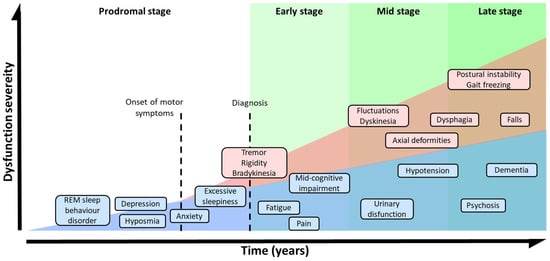
Figure 2. Symptomatic progression of PD. Schematic overview of both, MS and NMS symptoms progression and variability. PD diagnosis is based in MS, but NMS usually appears years before MS could be appreciated during the prodromal stage. The severity of the symptoms results from a combination of NMS, MS, and L-DOPA-derived complications.
3. Risk Factors and Genetics of Parkinson’s Disease
Neurodegenerative disorders constitute a diverse group of pathologies, with many challenges that need to be faced. Identifying the leading cause of the disease onset and progression is one of the major pending questions, and PD is a paradigmatic example. Although the intrinsic cause of PD onset and development remains uncertain, numerous factors (from environmental to genetic ones) have been reported to play a role. In the case of environmental factors, meta-analyses of case–control sets have described environmental factors as both increasing (pesticide exposure, prior head injury, rural living, β-blocker use, agricultural activities, and well-water consumption) and decreasing (tobacco, coffee, non-steroidal anti-inflammatory drug, and calcium channel blocker consumption and alcohol abuse) the risk elements for developing PD [78].
Even though aging is still considered to be the major risk factor, genetic studies have revealed that defects in numerous genes also play a significant role in the onset and evolution of PD. The genetic alterations commonly associated with familial cases of the pathology have been summarized in Table 1. The first gene linked to PD was SNCA, which encodes for α-Syn and includes numerous single-point mutations. The first described SNCA mutation, A53T [40], was initially detected in patients that presented an accelerated course of the pathology, manifesting cognitive impairment after 5–7 years of onset and an average onset of 46 years [79,80]. One year later, researchers observed another single-point mutation: A30P [39], whose clinical profile, interestingly, revealed a more benign course of the disorder and a later onset. In the case of the E46K mutation [41], patients develop severe symptoms between the ages of 50 and 65, including dementia and autonomic failure [41,81]. Another genetic mutation, H50Q [43], was first described in patients that reported motor symptoms at the age of 60 and fast development of the pathology [43]. In the last decade, two other genetic mutations, G51D and A53E [44,82], have been reported to cause dementia and autonomic dysfunction and early motor symptom progression, respectively [44,83,84,85,86,87]. Furthermore, other single-point mutations, such as E83Q and A30G, which enhance α-Syn aggregation, have recently been described in PD patients [88,89]. SNCA duplications and triplications have also been observed in inherited PD. In this case, the disorder’s severity and progression are related to the number of gene copies, resulting in more severe symptomatology and early onset in patients affected by triplications of SNCA than those affected by duplications [42].
Table 1. The genetics of PD. Summary of the different gene involved in PD and their role in the development of the pathology.
| Gene | Role in PD |
|---|---|
| SNCA (α-Synuclein) | Protein aggregation Prion-like transmission Synaptic function and dopamine transmission |
| GBA (Glucocerebrosidase) | Lysosome mediated autophagy pathway |
| LRRK2 | Neurite structure Protein and membrane trafficking Lysosome-mediated autophagy pathway Synaptic function and dopamine transmission |
| MAPT (Tau) | Protein aggregation Neurite structure |
| VPS35 | Protein and membrane trafficking Lysosome-mediated autophagy pathway |
| DNAJC13 (REM-8) | Protein and membrane trafficking Lysosome-mediated autophagy pathway |
| GAK | Protein and membrane trafficking |
| RAB7L1 | Protein and membrane trafficking |
| RAB39B | Protein and membrane trafficking |
| Parkin | Ubiquitin-mediated proteasome Mitochondrial dysfunction and mitophagy |
| FBX07 | Ubiquitin-mediated proteasome |
| SCA3 (Ataxin-3) | Ubiquitin-mediated proteasome |
| PINK1 | Mitochondrial dysfunction and mitophagy |
| DJ-1 | Mitochondrial dysfunction and mitophagy |
| CHCHD2 | Mitochondrial dysfunction and mitophagy |
| POLG1 | Mitochondrial dysfunction and mitophagy |
| SREVF1 | Mitochondrial dysfunction and mitophagy |
| ATP12A2 | Lysosome-mediated autophagy pathway |
| SCARB2 (LIMP-2) | Lysosome-mediated autophagy pathway |
| SYNJ1 (Synaptojanin 1) | Synaptic function and dopamine transmission |
| GCH1 | Synaptic function and dopamine transmission |
| STX1B (Syntaxin-1B) | Synaptic function and dopamine transmission |
Other autosomal dominant mutations described in PD include genes with diverse functions. For example, missense mutations in the LRRK2 gene, which encodes for a large kinase with various functions such as vesicle trafficking or GTPase activity [90,91], have been identified in PD patients worldwide [49,92,93]. The mutant LRRK2 induces apoptotic neuroblastoma and cortical neurons death probably by altering the autophagy process [94,95]. Nevertheless, the most common genetic alterations, with more than 300 different mutations described, are observed in the GBA gene [96,97,98], which encodes for a lysosomal enzyme called glucocerebrosidase that degrades glucosylceramide into glucose and ceramide [99,100,101]. The role of GBA mutations in PD onset and progression is still under debate. Theories such as impaired lysosomal function or endoplasmic reticulum-associated stress have been related to GBA mutations in PD, but the accumulation of α-Syn is considered to be the most plausible hypothesis [102,103]. GBA carrier patients exhibit an early onset of PD with an acute motor deficit, but these mutations notably increase the severity of NMS, enhancing cognition impairment, depression, and anxiety, among other symptoms [97].
Although autosomal dominant mutations are the most common genetic factors of PD, autosomal recessive alterations have also been related to this pathology. The parkin gene, which encodes for an E3 protein–ubiquitin ligase, is one of these recessive mutants, including an altered number of gene copies and missense and nonsense mutations [104]. Parkin ligase regulates the degradation of misfolded proteins through the ubiquitin–proteasome system [105] and interacts with LRRK2 [106]. The role of these mutations in the development of the disease seems to relate to the accumulation of damaged mitochondria [107]. DJ1 and PINK1 mutations present similar features to those of parkin, as they participate in common biochemical pathways [45,107,108,109,110], differing in the presence of diffuse or complete LBs and Lewy neurites (LNs), respectively. On the one hand, mutations of DJ1, a modulator of gene expression under cellular stress [111,112,113], induce DJ1 migration to the outer mitochondrial membrane, increasing the sensitivity to stress [111,112]. On the other hand, PINK1 gene alterations include missense, nonsense, and splice mutations or small deletions or insertions [114,115] that induce a mitochondrial deficit and alter the mitophagy pathways [47].
4. Molecular Mechanism Implicated in PD Development
During PD progression, cells suffer numerous interconnected and retrograde alterations that culminate in the degeneration of specific brain regions (Figure 3). α-Syn seems to be a critical element around which most molecular and genetic factors converge [116,117,118,119]. In pathogenic conditions, α-Syn forms neurotoxic oligomeric structures that progressively assemble into insoluble fibrils and accumulate in LBs [21,116,117]. Recent studies have demonstrated that these aggregates could be transmitted to neighboring cells, seeding the aggregation of the protein in healthy neurons, and spreading the disease to different brain regions [120,121,122,123].
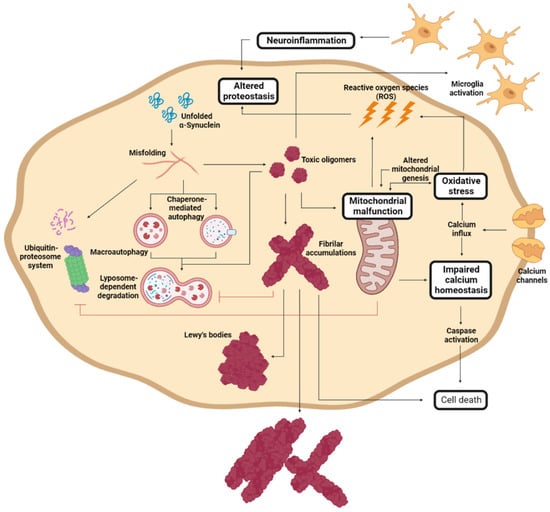
Figure 3. Molecular mechanism of Parkinson’s disease cell damage. Schematic representation of the interconnected molecular processes that induce cell death and PD progression. Adapted from [61].
α-Syn proteostasis depends on the ubiquitin–proteasome and the lysosomal autophagic systems [119,124], whose inhibition leads to the accumulation of α-Syn [118,125]. Accordingly, mutations in the LRRK2, GBA, and/or VPS35 genes translate into a pronounced number of LBs and LNs [101,126,127,128]. Conversely, the pharmacological stimulation of autophagy systems significantly decreased the aggregated α-Syn in animal models [129,130]. Oligomeric α-Syn and its accumulation alter the function of the ubiquitin–proteasome system, inhibiting macroautophagic processes and chaperone-mediated autophagy [131].
The aggregation of α-Syn also induces mitochondrial dysfunction, which stimulates the formation of amyloid fibrils [94]. α-Syn accumulation decreases the levels of peroxisome proliferator-activated receptor-γ co-activator 1α, a mitochondrial regulator of the transcription [132,133], which reduces the levels and toxicity of α-Syn oligomers when it is activated [134]. This relates to the relevance of LRRK2 (mutations leading to mitochondrial impairment), Parkin, and PINK1 (responsible for the degradation of harmed mitochondria) in PD development [94,110]. As a result of mitochondrial dysfunction, there is a significant accumulation of metabolites that produce high oxidative stress levels [135], to which unmyelinated DA neurons are susceptible [136,137]. Mutations of DJ1 significantly reduce the cellular response to stress [108,111,112]. Mitochondrial deficiency also impacts the energy levels of the neuron, inducing impaired calcium homeostasis and rapid axonal degeneration [138,139].
Another molecular process that plays an important role in PD and that is closely related to α-Syn aggregation and spreading involves immune system activation [140]. α-Syn aggregation stimulates an adaptative and innate immune response to toxic amyloids, but, at the same time, the generated neuroinflammation retroactively enhances protein aggregation [141,142,143], aggravating neuroinflammation. Therefore, modulating the immune response to toxic α-Syn species has been extensively studied as a therapeutic alternative.
5. Alpha-Synuclein
Due to the connection between α-Syn and PD onset and progression, this protein has become the preferred target in the search for a disease-modifying treatment [56]. α-Syn is a 140 amino acid protein encoded in SNCA gene and is mainly expressed in the brain’s synaptic termination of DA neurons. In normal conditions, α-Syn can be found as a soluble, monomeric, and disordered protein or bound to lipidic membranes, with an alpha-helical conformation (Figure 4B) [144]. Some studies have also revealed the possibility that α-Syn forms a tetrameric and helical structure in the cytoplasm [145]. Although its function remains unclear, it has been related to vesicle trafficking at the synapsis [146], participating in the release and recycling processes. This activity might be mediated by its interaction with VAMP2, a synaptobrevin involved in the fusion and binding of synaptic vesicles [147]. This interaction stabilizes SNARE complexes, which intervene in vesicle fusion and neurotransmitter release [148,149]. Nevertheless, α-Syn might also play alternative functions. For example, some studies suggest that α-Syn stabilizes the mRNA in P-bodies by binding proteins found at these membrane-less organelles [150], while others suggest that α-Syn may modulate DNA repair [151].
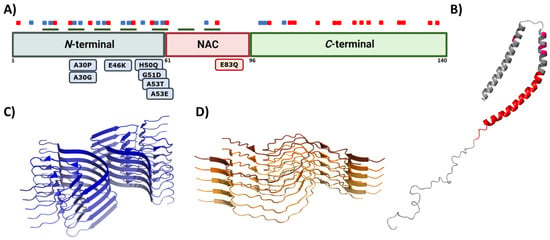
Figure 4. Alpha synuclein architecture. (A) Schematic representation of α-Syn primary sequence indicating the location of positively (blue) and negatively (red) charged amino acid, and KTKEGV repeats (green). Sequential domains and single-point mutations related to familial cases of PD are also indicated below the linear representation. (B–D) Structure of monomeric (B) and aggregated α-Syn forming different conformations or strains (C,D). In (B–D), the PDB files used are: 1XQ8, 6CU7, and 6CU8, respectively.
This multivalent activity is connected to its particular sequence, which could be dissected into three different regions (Figure 4A) [152]. The N-terminal domain is a highly conserved protein region that concentrates the most imperfect KTKEGV repeats. These repeats confer an amphipathic character responsible for the conformational change to an α-helical configuration and the protein–lipid interaction that dictates the binding to the membranes [144,153]. Significantly, this protein–lipid interaction has been described as a risk factor when the concentration of α-Syn increases, as it facilitates local nucleation for amyloid formation [154,155,156,157]. Recent studies have also proved that the N-terminal region contains two sequences (residues 36–42 and residues 45–57) with an essential role in homomeric α-Syn interactions, establishing contacts between monomeric α-Syn proteins that precede cross-β formation [158,159,160]. In addition, most of the missense mutations related to the early onset of PD, MSA, or DLB are also found in this region [38,39,40,41,43,44]. The central region is also known as the Non-Amyloid Component (NAC), as it is an important component of amyloid plaques in AD [161]. This is a hydrophobic segment often protected by the transient interactions that occur because of the disordered nature of the protein, but it drives the aggregation of α-Syn in pathogenic conditions [25,162,163,164]. In contrast, the C-terminal domain presents a large amount of acidic amino acids that provide a highly negative charge density. This net charge seems to chaperone α-Syn aggregation by electrostatic repulsions [165]. Accordingly, C-terminal truncations of α-Syn increase the aggregation propensity and toxicity and are important components of LBs, which suggests that this process could play a relevant role in pathogenesis [24].
In normal conditions, α-Syn displays significant solubility, but in pathogenic situations, this protein tends to establish β-sheet interactions that induce the formation of insoluble amyloid-toxic structures, compromising cellular homeostasis and inducing neuronal death [166], as outlined above. Amyloid aggregation is a complex process that, in vitro, often can be assimilated to a sigmoidal representation with three different phases, reflecting a nucleation–polymerization process [167]. During the lag phase, α-Syn monomers interact, forming toxic and transmissible structures named oligomers and protofibrils [168] that will act as the aggregation nuclei. The second step, or the elongation phase, consists of an exponential increase in the number and size of the fibrils. Finally, the plateau phase is characterized by the presence of mature and long amyloid fibrils (Figure 5). However, this is a very simplified description of the aggregation process, which in addition comprises alternative events caused by fibril fragmentation (secondary nucleation) or seeding processes, which contribute to accelerate or abrogate the nucleation phase (Figure 5) [169].
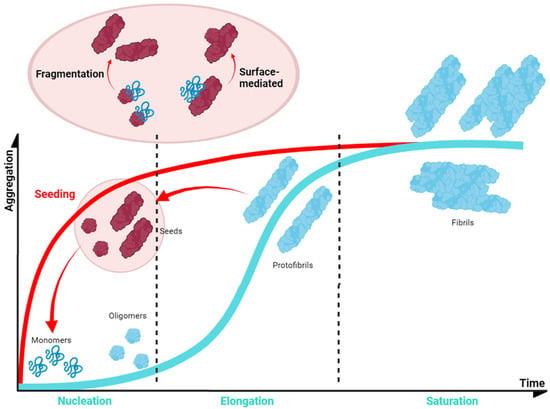
Figure 5. Schematic α-Syn aggregation profile. The aggregation kinetics of most of the proteins can be dissected into three main phases (blue). This also applies to α-Syn, for which the first step (nucleation or lag phase) is characterised by the formation of small nucleus that would guide the process; these nuclei incorporate monomeric protein prompting an exponential growth of the aggregate (elongation or exponential phase); finally, the system enters in an equilibrium in which mature fibrils could be observed (saturation or plateau phase). However, the process could be accelerated (red) as fibrils can fragment into smaller aggregates that can be incorporated at the initial stages as nuclei or seeds (seeding).
Genetic and environmental factors further modulate the aggregation process of α-Syn. Missense mutations of α-Syn (A30P, E46K, H50Q, G51D, A53E, and A53T), which are located at the membrane-binding region (Figure 4A) and impact the protein aggregation propensity, inducing either its oligomerization (A30P, H50Q, and A53T) or fibril formation (H50Q, A53T, E46K, and E83Q), and, thus, the formation of α-Syn toxic species [170]. In contrast, G51D and A53E variants slow down α-Syn aggregation compared to that of wild-type (WT) α-Syn, but they alter its interaction with the membranes [82,171]. Despite the exact role of these mutants in PD is still unknown, patients suffering G51D and A53E mutations exhibit a large amount of α-Syn pathological inclusions (in the case of G51D, also in oligodendrocytes) [44] and an earlier onset of PD. Regarding this evidence, it is suggested that these missense mutations could prolong the lifetime or stimulate the generation of toxic α-Syn structures such as oligomers [82]. Moreover, recent studies have shown that environmental factors, such as ionic strength or pH, affect the intermolecular interactions of α-Syn and contribute to the heterogeneity of the aggregation process. The analysis of the effect of pH variations on α-Syn aggregation suggested that acidic conditions (ranging from pH = 5 to pH = 3) induce the formation of partially folded species containing the β-sheet conformation, while retaining the monomeric ones [172]. This partial folding, as reported in terms of Thioflavin-T (Th-T) kinetics, prompts a significant decrease in the lag phase, while increasing the elongation rate, and, thus, stimulating α-Syn aggregation probably by impacting the protonation state of the C-terminal domain [172]. Regarding ionic strength, several studies have been performed to elucidate the role of salt in α-Syn aggregation. On the one hand, when α-Syn is incubated in the presence of preformed fibril seeds (PFFs) or lipid vesicles, the seeded polymerization of the protein notably decreased with an increasing salt concentration [173]. In contrast, when monomeric α-Syn is aggregated in the absence of these seeds, the salt significantly promotes the aggregation of WT and familial variants of α-Syn and impacts the fibrillar structure [174,175,176]. Although both results may seem to be contradictory, they are, in fact, complementary. As reported in several studies [117,177,178], the presence of salt during the aggregation process compensates for the electrostatic repulsions exerted by the C-terminal region, thus precluding the anti-aggregational effect of this region and accelerating protein aggregation. Nevertheless, the fibrils obtained in the presence of salt presented a higher level of compaction than the ones formed in its absence do, which has been suggested to result from the hiding of the C-terminal acidic domain, which is normally exposed and forms a fuzzy coat. Interestingly, the presence of these disordered regions on the fibril might play a relevant role in seeded polymerization. When this region is exposed at the PFFs surface, it has been reported to promote the amyloid aggregation of α-Syn either in vitro or in cellular models. In these cellular models, the C-terminus facilitates PFFs internalization by interacting with cell surface receptors [117,177,178]. Accordingly, and as for human prion proteins [179,180], α-Syn fibrils formed under different solution conditions share a common cross-β fold, but exhibit different conformation, seeding activity, neurotoxicity, and spreading in cells and when they are inoculated in rat brains [177,181,182,183,184,185]. These diverse conformational assemblies are called strains (Figure 4C,D) and could explain the existence of different synucleinopathies with unique clinical features [182,186,187] as their different properties would induce particular lesion profiles and brain region dissemination [188]. Indeed, a recent Cryo-EM comparative study evidenced structural differences between α-Syn fibrils obtained from MSA patients and those obtained from patients with DLB [186], supporting the existence of functionally distinct strains in humans.
Recent studies have suggested that in addition to aggregation, α-Syn can undergo liquid–liquid phase separation (LLPS) in vitro and in vivo as a previous step to amyloid formation [189,190,191]. LLPS is a recently described aggregation-related phenomenon characterized by the formation of multivalent macromolecular interactions, which induce the formation of an alternative phase with particular physicochemical properties that may be the main responsible for the formation of membrane-less organelles [192,193,194,195,196,197]. The mechanism underlying the transition of LLPS to amyloid is still unclear, with only a few structural studies addressing it [191]. Environmental conditions, such as pH or ionic strength, also condition LLPS and the maturation into aggregates [198]. This might be the intrinsic mechanism that accounts for the formation of different strains. Remarkably, environmental risk factors for PD, such as the Ca2+ or Mn2+ cations, have been proven to facilitate LLPS and accelerate aggregation [199,200]. Thus, even though the functional role of α-Syn in LLPS remains unclear, understanding the interactions that lead to amyloid transition might permit the stabilization of LLPS and prevent further progress toward aggregation [201]. Moreover, new studies have suggested that α-Syn and other amyloid-like proteins, such as prion and tau, may be synergistically connected via LLPS with different amyloidosis [202,203].
In regard to in vivo studies, the formation of α-Syn fibrillar structures is preceded by the assembly of the monomeric protein into small and diffusible metastable oligomers and protofibrils, which have been suggested to be the main culprits of neuronal degeneration [168,204,205,206,207,208,209]. These aggregated, but diffusible, structures progressively appear in different brain regions [210], consistent with a prion-like transmission mechanism as proposed by the Braak’s theory. This hypothesis suggests the development of PD follows a sequential pattern, beginning in the dorsal motor nucleus of the vagus nerve in the brainstem, and then spreading to other areas of the brain, which would explain the gradual development of different PD symptoms [120,211].
6. New Therapies: Modulating α-Synuclein Aggregation
The lack of an effective therapy targeting PD’s molecular basis has led to a continuous search for new treatments. One of these approaches is gene therapy, which has become a relevant strategy for treating numerous diseases. Lentiviral and adeno-associated viral vectors that have been approved for human use [212,213,214] are the most studied in PD, with different targets being identified for possible gene treatments, including disease modifiers and non-modifiers. Glutamic acid decarboxylase (GAD) overexpression through adeno-associated vectors administration was the first gene therapy studied in PD patients. It improved their symptomatic profile, but did not result in neuroprotective activity [215]. Other studied therapies are based on L-amino acid decarboxylase gene administration, alone or combined with tyrosine hydroxylase and GTP cyclohydroxylase 1 [216,217]. The L-amino acid decarboxylase gene plays a crucial role in dopamine metabolism, but genetic therapies targeting this gene only resulted in a Unified Parkinson’s Disease Rating Scale (UPDRS) score improvement [216,217]. Alternative gene therapies are based on the overexpression of growth and/or neuroprotective factors, such as glial cell-line derived neurotrophic factor, neurturin, artemin, persephin, vascular endothelial growth factor, or Nurr1 [218,219,220,221]. It has also been suggested that CRISPR/CAS9 technology could be used to correct genetic mutations associated with PD [222]. Other studied approaches were based on cellular transplantation. During the 1990s, fetal cell transplantation capacity to restore striatal dopamine transmission and connectivity and MS improvement was investigated [223,224]. Although the first results suggested that it could induce side effects [225], advances in stem cells, which can develop into DA neurons after grafting in animal models, might provide a new opportunity for cell transplantation as a therapy for PD [225,226]. Nonetheless, α-Syn aggregation has become the most investigated target in the search for putative therapies for PD. Different strategies have been explored, including SNCA gene-silencing to reduce the neuronal levels of α-Syn, strategies to increase the clearance of aggregated α-Syn by stimulating autophagic or proteasomal activities, and agents that prevent the formation and/or spreading of toxic aggregated structures [56,227]. Antibodies, vaccines, molecular chaperones, and small molecules are some of the most representative agents to target α-Syn aggregation in CNS and PNS. The difficulty for protein-based drugs to cross the BBB and the possibility of developing collateral immunological reactions make small molecules one of the preferred options in PD drug development [228,229].6.1. Polyphenolic Scaffolds
The aggregation of α-Syn is a complex process that involves different conformations that small molecules could target to interfere with this process (Figure 6). Accordingly, there are many chemically diverse compounds discovered by different methodologies, which exhibit different mechanisms of action to target α-Syn aggregation (Figure 7).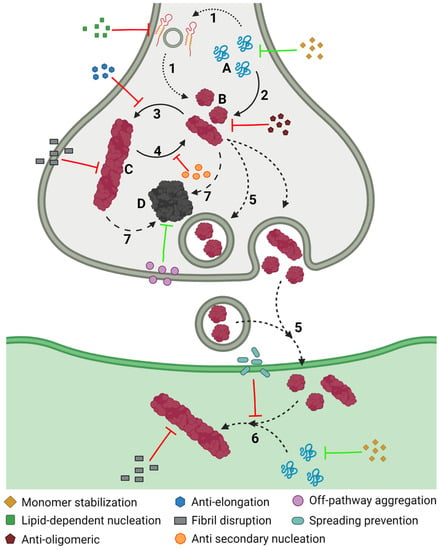
Figure 6. Inhibiting α-Syn aggregation. Schematic representation of the different mechanisms, illustrated by coloured symbols, available to prevent the aggregation of α-Syn, which follows a slow process that comprises different assemblies of the protein: monomeric (A), oligomeric (B), and fibrillar (C), or amorphous (D) aggregates. This process comprises different steps during the development of PD: protein–lipid interaction (1), oligomerization (2), fibril elongation (3), secondary nucleation (4), transmission (5), seeding (6), and amorphous aggregation (7).
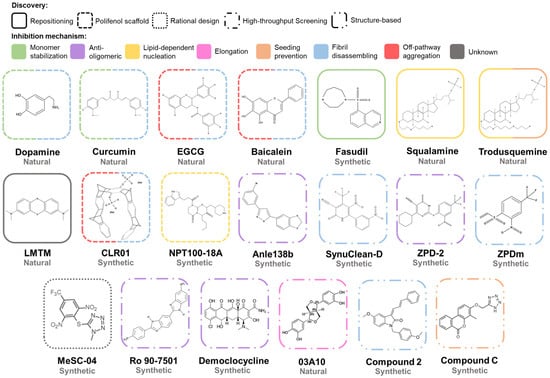
Figure 7. Chemical structures of inhibitors of α-Syn aggregation. Molecular structures of the most relevant modulators grouped by molecular class and mechanism of action. Abbreviations: EGCG, epigallocatechin-3-gallate; LMTM, leuco-methylthioninium bis(hydromethanesulphonate).