Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Hasniza Zaman Huri and Version 2 by Camila Xu.
Type 2 diabetes mellitus (T2DM) is characterized by impaired insulin secretion on a background of insulin resistance (IR). IR and T2DM are associated with atherosclerotic coronary artery disease (CAD). The mechanisms of IR and atherosclerosis are known to share similar genetic and environmental roots.
- insulin resistance
- type 2 diabetes mellitus
- atherosclerosis
- endothelial dysfunction
1. Background
Type 2 diabetes mellitus (T2DM) accounts for over 90 per cent of all patients with diabetes [1]. T2DM shares several risk factors in common with coronary artery diseases (CAD), such as aging, hypertension, dyslipidemia, obesity, lack of physical activity, genetics, and stress. In addition, an increase in the prevalence of diabetes indirectly escalates the risk of CAD [2]. T2DM is primarily caused by insulin resistance (IR), in which insulin cannot promote glucose uptake in skeletal muscle and adipose tissue and suppress hepatic glucose output [3][4][3,4].
Endothelial dysfunction (ED), the failure of endothelium to maintain vascular homeostasis, is present at the early stages of IR. It may be the origin of the initiation and progression of atherosclerosis [5]. Atherosclerosis that affects the coronary arteries can cause coronary artery disease (CAD) [6]. CAD is a type of cardiovascular disease (CVD) that is often asymptomatic in T2DM patients until the onset of myocardial infarction (MI) or sudden cardiac death [7].
IR may be associated with ED via various mechanisms such as disturbances of the subcellular signaling pathways that involve insulin action and nitric oxide (NO) production, oxidative stress, endothelin, the secretion of hormones and cytokines, as well as the renin–angiotensin–aldosterone system [8]. Changes in the subcellular signaling pathways convert the anti-atherogenic property of NO into pro-atherogenic and cause the development of atherosclerosis and subsequently CAD [9].
ED has been linked to several factors such as diabetes, hypertension, smoking, a high-fat diet, as well genetic factors [10]. The genetic factor of ED involves the dysregulation of the endothelial NO synthase (eNOS) gene followed by disruption of the endothelial vascular homeostasis [8]. It has been reported that the polymorphism caused by four or five repeats of a 27-base-pair sequence in intron 4 of the eNOS gene is associated with the risk of CAD [11]. Another study of the eNOS gene also found that intron 4 polymorphism was associated with T2DM [12]. Thus, the genetic factor of ED is linked to both T2DM and CAD [13].
The eNOS gene expression pathways involve the activation of the pro-inflammatory markers related to IR, atherosclerotic CAD and T2DM [14]. These genes and their single-nucleotide polymorphisms (SNPs) may act as the possible identifiers of IR and atherosclerosis in T2DM patients with CAD.
2. How Does IR Result in T2DM?
Insulin is a peptide hormone secreted by the β cells of the pancreatic islets of Langerhans. It maintains normal blood glucose levels by facilitating cellular glucose uptake, regulating carbohydrate, lipid, and protein metabolism, and promoting cell division and growth through its mitogenic effects [15]. Insulin secretion may be influenced by alterations in synthesis at the level of gene transcription, translation, and post-translational modification in the Golgi among other factors influencing insulin release from the secretory granules [15][16][15,16]. The actions of insulin are influenced by the interplay of other hormones such as growth hormone, IGF-1, glucagon, glucocorticoids, and catecholamines [17]. Excessive hormone production might have contributed to IR in some circumstances, though com-promised insulin signaling is thought to have a greater role at the cellular level [18]. As such, IR can be considered as a state of chronic, low-level inflammation [19]. Several states or mechanisms result in IR, such as fatty acid-induced IR [20] and lipid accumulation in skeletal muscle and liver [21]. The beginning of IR frequently results in insulin paucity and a gradually dwindling blood glucose regulation, hyperinsulinemia, and increased levels of free fatty acid (FFA) circulation [22][23][22,23]. The circulating FFAs are the main substrate for production of hepatic triglycerides in the form of very low-density lipoprotein (VLDL) [24][25][26][24,25,26]. These changes are trailed by a subsequent decline in plasma glucose control, which manifests as elevated fasting plasma glucose levels with intermittent and persistent hyperglycemia leading to a diagnosis of T2DM (Figure 1) [27]. Therefore, in terms of pathogenesis, glucolipotoxicity is stated as an essential determinant of T2DM. Glucolipotoxicity is the combination of glucotoxicity (hyperglycemia or elevated blood glucose levels) and lipotoxicity (high lipid levels, especially FFAs) [28].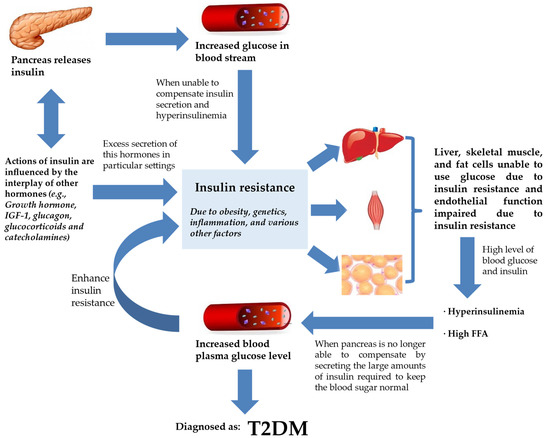
Figure 1. IR and T2DM. Insulin secretion by the pancreas is influenced by the interplay of various hormones (hormone, IGF-1, glucagon, glucocorticoids, and catecholamines) in order to regulate glucose levels in the bloodstream. However, insulin failure to manage blood glucose will lead to the progression of IR. IR will compromise endothelial function, rendering liver, skeletal muscle, and fat cells unable to use glucose. This worsens hyperinsulinemia, and the pancreas is already incapable of keeping normal blood glucose despite secreting large amounts of insulin. Blood plasma glucose levels keep increasing and the feedback loop enhances IR further. At this stage, the patient is diagnosed with T2DM. Abbreviations: ED, endothelial dysfunction; FFA, free fatty acid; IGF-1, insulin-like growth factor 1; IR, insulin resistance; T2DM, type 2 diabetes mellitus.
3. Atherosclerosis and Its Relationship with IR, T2DM, and CAD
T2DM and atherosclerotic CVD share many common factors. In T2DM, inflammatory processes play a part in the cause of atherosclerotic CVD [34]. The markers of inflammation predict CAD and the levels of markers are raised in patients with T2DM [35]. ED is considered as an early marker for atherosclerosis, and it plays a prominent role in the development of IR and T2DM [5]. IR and impaired insulin secretion are central to the pathogenesis of T2DM, but it is unclear how these abnormalities are related to accelerated atherosclerosis [36]. The precise mechanisms for the susceptibility and advancement of atherosclerosis in patients with T2DM are undetermined. Yet, there are many indications linking lipoproteins with atherosclerosis [37]. Lipoproteins are the combinations of fats and protein. The lipoproteins are proved to play an important role in atherosclerosis, and they interact with the artery wall to trigger the chain of events that leads to atherosclerosis. Low-density lipoprotein (LDL) is the most common lipoprotein that is associated with atherosclerosis, but other lipoproteins such as VLDL may also be atherogenic. An important component of these atherogenic lipoproteins is Apolipoprotein B (Apo B) that promotes the accumulation of LDL in the intima initiates atherosclerosis [38]. This is mediated by increased endothelial permeability and raised intimal retention of LDL [39]. Moreover, diabetes (T1DM and T2DM) is associated with increased hepatic production of triglyceride-rich lipoproteins, which leads to increased formation of atherogenic VLDL [40]. The atherogenic index is defined as the ratio of LDL-C (low-density lipoprotein cholesterol) to HDL-C (high-density lipoprotein cholesterol). In one study, researchers discovered that hyperlipidemic rats had a higher atherogenic ratio (8.06 mg/dL) than the control group (1.09 mg/dL) [41]. Metabolic syndrome, pre-diabetes, and T2DM, which all co-segregate with IR, accelerate the progression of atherosclerosis and the consequential disease [34]. Silent atherosclerosis and cardiovascular complications start to commence during the pre-diabetic period in genetically susceptible people [42]. During the onset of diabetes, hyperglycemia and hyperinsulinemia are present in IR and lead to acceleration of atherosclerosis and CAD (Figure 2) [43][44][43,44]. Still, despite the fact that diabetic patients developed severe lesion formation, patients with and without diabetes often have identical atherosclerotic lesions [36]. Areas of necrotic lesions accumulate lipid, cholesterol crystals, and inflammatory cells, which leads to thickening of the arterial wall. Complications can arise from erosion or calcification, causing a thrombus to form and obstruct the lumen, resulting in health defects such as CAD [8][44][8,44].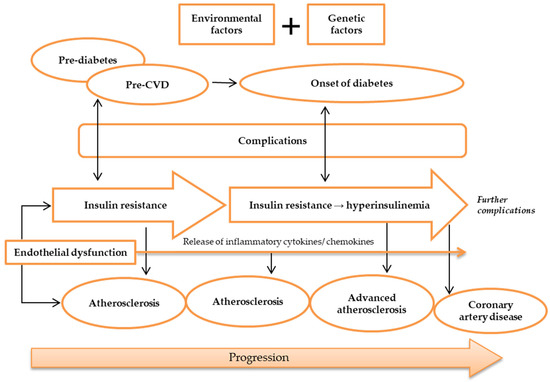
Figure 2. Pathogenesis of atherosclerosis. Atherosclerosis may be caused by environmental and genetic factors. The onset of atherosclerosis is during ED initial occurrence, which is also associated with the onset of IR. These incidences reflect pre-diabetes and pre-CVD phases, and symptoms of complications may appear. Ongoing atherosclerosis development is incessantly supported by ED, linked to the release and regulation of inflammatory cytokines and chemokines, as well as IR, which develops into hyperinsulinemia. At this stage, patients may have developed advanced atherosclerosis and diabetes, while exhibiting evident complications. Further ED, IR, and atherosclerosis advancement will result in more apparent complications and CAD development. Abbreviations: CAD, coronary artery disease; CVD, cardiovascular disease; ED, endothelial dysfunction; IR, insulin resistance.