The widespread use of peptide receptor radionuclide therapy (PRRT) represents a major therapeutic breakthrough in nuclear medicine, particularly since the introduction of 177Lu-radiolabeled somatostatin analogs. These radiopharmaceuticals have especially improved progression-free survival and quality of life in patients with inoperable metastatic gastroenteropancreatic neuroendocrine tumors expressing somatostatin receptors. In the case of aggressive or resistant disease, the use of somatostatin derivatives radiolabeled with an alpha-emitter could provide a promising alternative. Among the currently available alpha-emitting radioelements, actinium-225 has emerged as the most suitable candidate, especially regarding its physical and radiochemical properties.
- actinium-225
- radionuclide production
- radiolabeling
- targeted radionuclide therapy
- targeted alpha-therapy
- radiobiology
- neuroendocrine tumors
- radiopharmaceuticals
- 225Ac–DOTATATE
- 225Ac–DOTATATE
1. Introduction
1.1. About Neuroendocrine Tumors
1.2. Somatostatin Receptors and Octreotide Analogs
Although NETs are heterogeneous diseases in their pathophysiology and clinical expression, they usually share the characteristic of overexpressing somatostatin receptors (SSTRs) [4][13]. Five SSTR subtypes are described (SSTR1 to SSTR5), SSTR2 being the most frequently encountered in differentiated NETs [5][14]. However, several subtypes can be expressed concomitantly on tumor cells in various combinations and proportions [6][7][15,16]. NETs overexpressing SSTRs most often have a gastrointestinal, pancreatic, bronchial, pulmonary, or even thymic or breast origin. SSTRs belong to the G-protein-coupled receptor family and are localized at the cell membrane. Their natural peptide ligand, somatostatin, is found in humans under two different forms: one of 14 amino acids (SS-14) and one of 28 amino acids (SS-28) (Figure 1) [8][9][17,18]. Natural somatostatin has been shown to be unsuitable for in vivo use due to its short plasma half-life (about 3 min) [10][19]. Analogues of this hormone, more resistant to enzymatic degradation, have therefore been developed by making various modifications to the natural molecule [11][12][20,21]. The introduction of D-series amino acids to improve in vivo stability, the retention of the minimum chain length to maintain biological activity, the use of the hexapeptide motif Cys-Phe-D-Trp-Lys-Thr-Cys and the elongation of the N- and C-terminal ends allowed the characterization, in 1982, of the most stable active somatostatin analog known as octreotide (Figure 1) [13][22].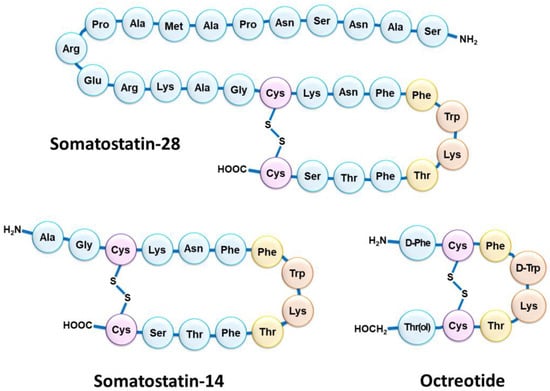
1.3. SSTR Targeting for Peptide Receptor Radionuclide Therapy
1.4. PRRT Using Somatostatin Analogs Radiolabeled with Alpha-Emitters
Although β-PRRT remains an approved treatment for unresectable metastatic NETs, some tumors show resistance to β-emissions despite somatostatin receptor expression [25][69]. Furthermore, not all treated patients achieve partial or complete response following SSTR-targeting 177Lu-PRRT, and relapse is often observed in the years post-treatment [26][70]. Thus, among the strategies considered in an effort to overcome these drawbacks, octreotide derivatives radiolabeled with alpha-emitting radionuclides have received particular attention [27][71]. Within this group of radioisotopes, radium-223 (alkaline earth metal, group 2) has been extensively studied both in vitro and in vivo, and has paved the way for the use of alpha-emitting radioelements in patients [28][72]. To date, radium-223 is used in its dichloride form for the treatment of symptomatic bone metastases in patients with castration-resistant prostate cancer, without known visceral metastatic disease. However, due to its particular chemistry, 223Ra is not suitable for DOTA-peptide radiolabeling. Thus, a special interest has emerged for several α-emitting lanthanides (e.g., 149Tb) and actinides (e.g., 227Th and 225Ac), as well as some radioelements from their decay chain (e.g., 213Bi) to achieve a convenient complex formation with DOTA [29][73]. An initial preclinical evaluation of 213Bi–DOTATOC showed its potential value in NETs resistant to 177Lu-PRRT [30][31][32][33][74,75,76,77], these properties being promptly confirmed in the clinic [34][78].2. Actinium-225: Decay Characteristics, Radiobiological and Dosimetry Considerations
2.1. Physical Properties of Actinium-225
Actinium-225 is a relatively long-lived pure alpha-emitter, with a half-life of 9.9 days that is well-suited for radionuclide therapy applications and for centralized industrial production, distant from the (pre)clinical user sites. It is formed from the 229Th decay product 225Ra and decays via a cascade of six short-lived daughter radionuclides to the nearly stable bismuth-209 (Figure 23) [35][81]. These intermediates include francium-221 (t1/2 = 4.8 min, 6.3 MeV α-particle and 218 keV γ-emission), astatine-217 (t1/2 = 33 ms, 7.1 MeV α-particle), bismuth-213 (t1/2 = 45.6 min, 5.9 MeV α-particle, 1.4 MeV β-particle and 440 keV γ-emission), polonium-213 (t1/2 = 4.3 μs, 8.5 MeV α-particle), thallium-209 (t1/2 = 2.2 min, 3.9 MeV β-particle) and lead-209 (t1/2 = 3.2 h, 0.6 MeV β-particle) before reaching 209Bi. Overall, the predominant decay pathway of 225Ac produces four alpha-particles with energies ranging from 5.8 to 8.5 MeV and associated tissue ranges of 47 to 85 µm. In addition, the cascade includes two main beta-disintegrations of 1.4 and 0.6 MeV maximum energy. Therefore, 225Ac is considered as an in vivo radionuclide generator or a “nanogenerator” with regard to its decay chain.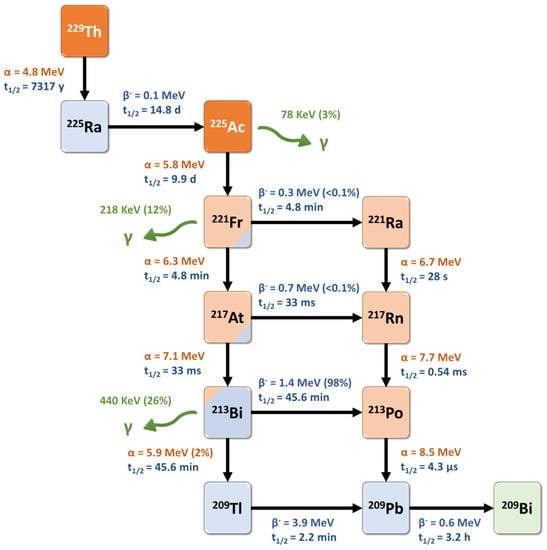
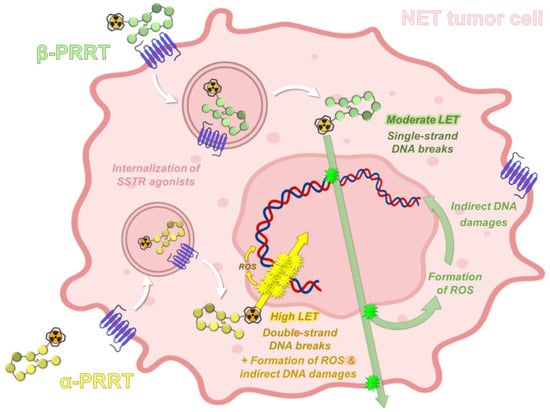
2.2. Radiobiological Properties of Actinium-225
2.3. Dosimetry for Targeted Alpha-Therapy with
225
Ac
3. Radiochemical and Preclinical Development of [225Ac]Ac–DOTATATE
3.1. Production of Actinium-225
Two isotopes of actinium, 227Ac and 228Ac, exist in nature within the natural decay chain of uranium-235 and thorium-232, respectively [44][94]. However, neither of these two isotopes is used in the clinic, with 228Ac representing a minimal part of natural actinium and 227Ac having a very long half-life (t1/2 = 21.77 y). Therefore, 225Ac is the only one of the more than 30 known actinium isotopes to be used in preclinical and clinical studies to date. The main method for generating 225Ac for clinical use is through radiochemical extraction following the decay of 229Th (t1/2 = 7397 y), which originates from reactor-bred 233U [45][46][95,96]. The main sources of 229Th in the world for 225Ac used in preclinical and clinical studies are Oak Ridge National Laboratory (ORNL, Oak Ridge, TN, USA) [45][95], the Institute of Physics and Power Engineering (IPPE, Obninsk, Russia) [47][97], and the Directorate for Nuclear Safety and Security of the Joint Research Center of the European commission (JRC, Halstenbek, Germany), formerly the Institute for Transuranium Elements (ITE, Karlsruhe, Germany) [48][98]. More recently, the Canadian Nuclear Laboratories set up a 225Ac production chain that could supply up to 3.7 GBq of this radioisotope annually [49][99]. Consequently, accelerator-based production techniques to obtain 225Ac have been developed. The most promising approach to obtain 225Ac at a large scale may be the cyclotron proton irradiation of a 226Ra target, involving the 226Ra(p,2n)225Ac transformation [50][51][111,112]. With a high cross-section peak (710 mb) at 16.8 MeV, this convenient method can be performed on low-energy cyclotrons.3.2. Chemistry of Actinium
3.2.1. Actinium in Aqueous Solution
Actinium is the chemical element with atomic number 89 and the first element of the actinide group, to which it gives its name. Nevertheless, actinium has rather similar chemical properties to lanthanum and other lanthanides. Actinium exists essentially in the +3 oxidation state in aqueous solution; additionally, Ac3+ is the largest +3 cation in the periodic table. It is also the most basic +3 ion due to its low charge density, directly related to its large size. Although the +3 state is the most stable in aqueous solution, the +2 oxidation state may also be encountered [52][120]. This second species is assumed because a reduction half-wave potential in a 225Ac3+ aqueous solution can be observed. The progressive negative shift of this potential in the presence of increasing 18-crown-6 concentrations has been attributed to the formation of a complex between crown ether and divalent actinium [53][121]. However, without the effect of 18-crown-6 on the reduction of 225Ac3+, the existence of stable 225Ac2+ ions in aqueous solution remains unlikely regarding the low extraction yields of actinium using sodium amalgam in aqueous sodium acetate, an extraction technique usually efficient for lanthanides at a stable +2 oxidation state [54][122].3.2.2. Coordination Chemistry of Actinium
The usefulness of actinium-225 as a radionuclide for therapeutic purposes has been limited for a long time by the unavailability of chelating agents that are both capable of being compatible with this bulky radionuclide and of controlling the fate of the resulting daughter emitters, particularly with regard to their alpha-recoil, which is related to the conservation of momentum laws that occurs upon release of an alpha-particle [55][137]. Nevertheless, the coordination chemistry of such a clinically relevant alpha-emitter has recently gained more and more interest [56][138]. Considering its low polarizability and despite its large ionic radius, the Ac3+ ion is considered a hard Lewis acid [57][139], showing a medium absolute chemical hardness value of 14.4 eV [58][140]. As such, it will complex more easily with hard ligands, such as anionic oxygen donors. The complexation reaction will preferentially occur under charge control and the acid–base bond will be essentially ionic. Indeed, Ac3+ displays an electrostatic interaction constant (EA) value of 2.84 and a covalent interaction constant (CA) value of 0.28 [56][138]. This predominance of charge interactions can be predicted from the character of the frontier molecular orbitals, which are centered on the nuclei of the donor and acceptor atoms; when these atoms are close together in space, the overlaps of the orbitals are negligible while the charge interactions are strong. This is mainly attributed to the density of the charge, which is very significant in ions of hard consistency. The high ionic radius of the Ac3+ cation suggests that the most suitable chelators would be polydentate agents, with a high denticity between 8 and 12. Initial works investigated the suitability of linear polyaminocarboxylate chelators, such as CHX-A″-DTPA, for the chelation of the 225Ac3+ cation [59][60][141,142]. These efforts were motivated by the advantageous radiolabeling kinetic properties of these ligands; however, the complexes obtained did not show sufficient in vivo stability. Subsequently, large macrocyclic chelators were considered and the 18-membered polyaminocarboxylic acid core HEHA was rapidly identified as particularly suitable for actinium complexation [60][61][142,143].3.2.3. Relevance of DOTA in Actinium Radiopharmaceuticals
The in vivo fate of the 225Ac–DOTA complex alone was initially shown to be safe, with only low activity amounts in liver and bone of BALB/c mice [60][142]. Subsequently, DOTA-bioconjugated constructs (either antibodies or peptides) also showed the sufficient stability of the complex, both in vitro [62][63][64][65][162,166,167,173] and in vivo [62][63][162,166]. Nevertheless, early studies raised some concerns about the compatibility of DOTA with actinium [60][66][142,146]. Indeed, the large ionic radius of the Ac3+ ion is not in favor of the good thermodynamic stability of the DOTA complex, which may also be subject to transmetalation with other cations. In order to minimize adverse in vivo effects associated with the loss of 225Ac and its daughter radionuclides (especially 213Bi, significantly increasing the kidney-absorbed dose [67][194]) from DOTA, several approaches have been considered, such as the co-administration of chelating agents or concomitant diuresis [68][69][195,196].3.3. Somatostatin Analogs Radiolabeled with
225
Ac: Preclinical Studies
Only a few studies have reported preclinical efficacy results of 225Ac-radiolabeled somatostatin analogues, due to this group of vector molecules having already been widely studied with beta-emitters such as 90Y or 177Lu [70][197].
Activities between 10 and 60 kBq were well-tolerated by the mice; however, activities over 30 kBq induced pathologic changes in the renal cortex, suggesting radiation-induced acute tubular necrosis in both the distal and proximal tubules. Similar results were obtained in another study on Sprague Dawley rats that received 111 or 370 kBq [225Ac]Ac–DOTATOC and developed renal tubular nephrosis or renal glomerulopathy [71][199]. Only a slight accumulation in the liver was objectified, probably due to the release of free 225Ac. After a single administration of the highest non-toxic activity (20 kBq), tumor weights 14 days after treatment were lower with [225Ac]Ac–DOTATOC than with [177Lu]Lu–DOTATOC (1 MBq), in accordance with previous studies investigating [213Bi]Bi–DOTATOC [30][31][74,75].