Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Manuel Aureliano and Version 4 by Jessie Wu.
Lipid peroxidation (LPO), a process that affects human health, can be induced by exposure to vanadium salts and compounds. LPO is often exacerbated by oxidation stress, with some forms of vanadium providing protective effects. The LPO reaction involves the oxidation of the alkene bonds, primarily in polyunsaturated fatty acids, in a chain reaction to form radical and reactive oxygen species (ROS). The important question is which radical starts the chain first. On the one hand, a radical is needed (oxidative stress environment) while on the other hand, LPO amplifies and contributes to changing the redox state towards oxidation (what was called oxidative stress).
- lipid peroxidation
- vanadium
- oxidative stress
- cancer
- Parkinson
- Alzheimer
- Decavanadate
- Polyoxometalates
- Diabetes
- vanadate
- amyloid
1. Role of Vanadium in Lipid Peroxidation Related to Cancer
Despite somewhat controversial reports suggesting that vanadium may be an essential trace element for humans [1][2][165,166], pharmacological amounts of vanadium needed to observe efficacy may be 10 to 100 times the normal intake [3][167]. Under certain levels, some vanadium complexes/compounds have shown anticancer and/or antidiabetic activity in mammals [4][5][6][7][8][23,53,54,74,168], while higher levels can cause toxicity. Under some conditions, vanadium can act as a pro-oxidant molecule, which interacts with other oxidants and synergistically enhances oxidative stress and potentially lipid peroxidation (LPO) [9][11].
Reports from the early 1990s showed that V complexes/compounds induced LPO, which was associated with tissue toxicity and carcinogenicity [10][11][12][169,170,171]. Tissue-specific responses were shown for vanadate, which produced a cytotoxic response in the murine osteoblast-like MC3T3E1 nontransformed cell line [13][172]. This level of cytotoxicity was higher than that in vanadate-treated osteosarcoma cancer UMR106 cells with respect to both time- and concentration-dependent responses [13][172]. Osteoblastic cells were more sensitive to the vanadate-induced free radical and biomarker thiobarbituric acid (TBARS) formation, particularly at low concentrations. Nevertheless, higher basal TBARS was observed in untreated osteosarcoma cells [13][172]. Other vanadium compounds (VOSO4) and a complex of vanadyl with aspirin (VO/Aspi)) were found to be more potent than vanadate in inducing TBARS and inhibiting the cellular growth, in both cell lines tested [13][172] (Table 1). However, when an equivalent low concentration of VO/Aspi was released from a controlled delivery system (poly(β-propiolactone) (PβPL) film), less TBARS formation was observed [14][173] (Table 1), which reflects lower cytotoxicity compared to that previously reported for the metallodrug in solution [13][172].
The development and testing of vanadium derivatives with different ligands and with improved bioavailability and toxicity profiles continues. Both naproxen- and glucose-complexed vanadium compounds (NapVO and GluVO) had antiproliferative effects that were more pronounced in osteosarcoma UMR106 cells than in the normal MC3T3E1 osteoblasts [15][157]. This supported the observation that a low level of GluVO and NapVO increased TBARS production in tumoral cells but not in the nontransformed cells [15][157] (Table 1), suggesting LPO was involved in the antineoplastic action observed. Interestingly, neither the free vanadyl cation nor ligands induced an antimitogenic effect in cells at the concentrations tested [15][157]. At low concentrations, a large number of different complexes/compounds of vanadium were found to be therapeutically active [2][16][64,166]. Possible mechanisms for the anticancer activity of vanadium complexes/compounds included an increase in oxygen species (ROS) generation, hyperactivation of the Ras-Raf-MEK-ERK pathway and cell cycle arrest [17][18][19][174,175,176]. It is also possible that vanadium may confer protection against chemical-induced carcinogenesis or toxicity in normal tissues by normalization of increased pathogenic LPO and oxidative stress. While an increase in hepatic LPO was observed in a group of carcinogen-treated female Sprague Dawley rats, this increase was lowered towards normal values by vanadium co-administration [20][138] (Table 1) and was associated with a significantly lower percentage of rats with tumors after vanadium treatment. In these experimental groups, SOD activity in the liver paralleled LPO. By contrast, hepatic glutathione (GSH) and cytochrome P450 (CYP) enzyme content and glutathione S-transferase (GST) activity decreased with carcinogenic treatment compared to control rats and recovered with vanadium treatment [20][138]. Similarly, in a model of hepatocarcinogenesis induced in rats by chronic feeding of 2-acetylaminofluorene (2-AAF), continuous vanadium administration inhibited LPO and suppressed cell proliferation [21][177] (Table 12), suggesting vanadium was chemopreventive.
The chemoprotective role of vanadium against cancer chemotherapy-induced toxicity is also relevant. Many chemotherapeutic agents such as cyclophosphamide (CP) and cisplatin (CDDP) are toxic due to multifactorial mechanisms that include increased oxidative stress in normal tissues and organs, namely the liver and kidney. The co-administration of compounds with antioxidant potential may be beneficial to patients. For example, the simultaneous treatment of female Swiss albino mice with CP and either vanadium(III)-L-cysteine complex (VC-III) [22][178] or oxovanadium(IV)-L-cysteine methyl ester (VC-IV) [23][179] reduced ROS levels when compared to the increase in ROS observed in CP-treated group vs. control [22][23][178,179]. With respect to LPO, partial normalization of TBARS in CP/VC-III- or CP/VC-IV-treated mice was observed (Table 1) [22][23][178,179]. After treatment with CP, there was a decrease in GSH levels and in GST, glutathione peroxidase (GPx), superoxide dismutase (SOD) and catalase (CAT) activities, while a recovery was observed with vanadium treatment [22][23][178,179]. Similar protective effects were observed with concomitant treatment with cisplatin (CDDP) and VC-III (Table 12) [24][180]. These results suggest that vanadium may be beneficial as an adjunct therapy to protect against the toxicity of anticancer drugs.
Table 1. Effects of vanadium in lipid peroxidation related to cancer. Main outcomes of studies using different V compounds in various organs/tissues of animal models or cancer cells.
| Vanadium Compound | Combined/Complexed | Carcinogenic/Toxic Agent or Cell Lines | Tissue/Model | Main Results/Outcome | Ref. |
|---|---|---|---|---|---|
| V1V; VO | Aspirin; polymeric film |
Osteosarcoma UMR106 cells in culture | Bone | Cytotoxic effects | [13][14][172,173] |
| V1V derivatives | Naproxen (Nap-VO); Glucose (GluVO) | Apoptosis mediated by lipid peroxidation | [15][157] | ||
| Ammonium monovanadate (NH4VO3, +V oxidation state) (vanadium supplemented in drinking water) | 7,12-dimethylbenz(a)anthracene (DMBA)-induced mammary carcinogenesis in rats | Mammary gland | Prevention of mammary cancer | [20][138] | |
| Vanadium (in the form of ammonium vanadate) | 2-acetylaminofluorene (2-AAF)-induced hepatocarcinogenesis in rats | Liver | Vanadium was chemopreventive; inhibition of lipid peroxidation | [21][177] | |
| Oxovanadium(IV)-L-cysteine methyl ester (VC-IV) | Cyclophosphamide (CP)-induced hepatotoxicity in mice | Liver | Protective role of VC-IV against CP-induced toxicity | [23][179] | |
| Vanadium(III)-L-cysteine complex (VC-III) | Protective role of VC-III against CP- and CDDP-induced toxicity | [22][178] | |||
| Cisplatin (CDDP)-induced nephrotoxicity in mice | Kidney | [24][180] | |||
Even though vanadium participates in Fenton-type reactions [25][40] and the mechanisms proposed for vanadate action involve redox cycling and the production of ROS [26][27][65,66], some results show a depression in ROS and the rate of ROS formation [28][21]. Previous results show that in certain experimental conditions, for example in rats with induced hepatocarcinogenesis [29][137] and diabetes [20][138], vanadate may decrease oxidative stress. Strong evidence supports the observation that V10 alters the production of mitochondrial O2.− differently from V1 and suggests the possibility that different pathways are involved in the biological activity of different vanadium species. Of the proposed intracellular pathways for vanadate, several involve the production of O2.− mediated by oxidoreductases of NADPH in the respiratory chain [26][27][65,66]. Considering the proposed mechanisms of action and detoxification of vanadate, which include reducing vanadate to vanadyl with O2.− production, the available data support the interpretation that V10 may participate in bioprocessing and metabolism differently than V1.
2. Effect of Vanadium in Diabetes-Induced Lipid Peroxidation
Diabetes mellitus is a complex metabolic disease characterized by a chronic state of hyperglycemia [30][181]. Although the impaired function of the pancreatic islets might be relevant in its etiology, other tissues are affected and may present complications in uncontrolled disease [31][182]. Diabetes can be generally classified into different categories with distinct clinical features. Type 1 diabetes is an autoimmune disease in which beta cells in the pancreas are unable to produce the hormone insulin while in type 2 diabetes, the most common form of diabetes, the body is either resistant to insulin or incapable of producing sufficient amounts of insulin [30][181].
An imbalance between the production and removal of ROS and RNS may contribute to insulin resistance and pancreatic beta cell dysfunction, which ultimately leads to the development of type 2 diabetes [32][33][183,184]. Increased levels of TBARS, a biomarker of LPO, were more highly elevated in type 2 diabetes patients than in healthy control subjects [34][185]. In patients with type 2 diabetes, hyperglycemia was associated with increased oxidative stress and free radical-mediated LPO [35][36][37][186,187,188] both of which may affect the development of micro- and macrovascular complications related to the intensification of systemic inflammation in these patients [33][38][39][184,189,190]. Compounds that modulate LPO and oxidative stress and have an antioxidant potential may contribute to improving the metabolic health in patients with diabetes and be a valuable therapeutic approach.
In 1979, Tolman et al. showed that vanadium salts exhibited insulin-mimetic effects which led to an interest in vanadium chemistry for the treatment of diabetes [40][191]. Since then, a series of reports have been published describing the insulin-like effects of various vanadium compounds, mainly VIV and VV salt and coordination complexes. One coordination complex, an organic vanadium compound, bis(ethylmaltolato)oxovanadium(IV) (BEOV), exhibited excellent efficacy in streptozotocin (STZ)-diabetic rats [41][192] and entered Phase I and II clinical trials [42][43][193,194].
Using different animal models of diabetes and analyzing diverse tissues, many reports showed effects of vanadium compounds on the activity of antioxidant enzymes and the levels of LPO (Figure 1). Early studies from the 1990s showed that treatment of STZ-induced diabetic Sprague Dawley rats with sodium metavanadate (NaVO3), did not lead to changes in the antioxidant defense system [44][195]. However, the tissue level of vanadium positively correlated with the TBARS level [44][195]. By contrast, sodium orthovanadate (SOV) treatment of STZ-induced diabetic male Wistar rats led to the STZ-induced decrease in the hepatic activities of SOD, CAT and GPx being restored to normal levels, while the elevated plasma lipid peroxides (as measured by MDA) were decreased almost to basal values [45][196] (Figure 1). This same pattern was observed in the liver enzymes of alloxan-induced diabetes female Wistar rats (Figure 1), but not in all the tissues evaluated [29][137]. SOV treatment also almost normalized the chemical-induced increase in the levels of TBARS in the brain, along with the normalization of the activity of the brain GST, which was decreased in the diabetic rats [46][197] (Figure 1). Interestingly, subsequent studies have used SOV combined with Trigonella graecum seed powder (TSP) which makes it possible to use lower concentrations of vanadate. Most authors have shown a reversal of non-physiologic antioxidant levels and peroxidative stress in different tissues from diabetic animals [47][48][49][50][51][198,199,200,201,202] (Figure 1).
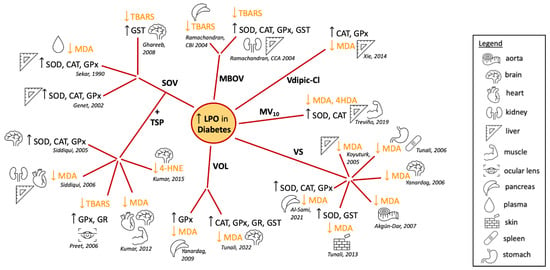
Figure 1. Summary of the reported effects of different vanadium compounds in lipid peroxidation and antioxidant enzyme activity, evaluated in different tissues of diabetes-induced animal models [29][45][46][47][48][49][50][51][52][53][54][55][56][57][58][59][60][61][62][63][137,196,197,198,199,200,201,202,203,204,205,206,207,208,209,210,211,212,213,214]. Abbreviations: superoxide dismutase, SOD; catalase, CAT; glutathione peroxidase, GPx; glutathione reductase, GR; glutathione S-transferase, GST; malondialdehyde, MDA; 4-hydroxy-2-nonenal, 4-HNE; thiobarbituric acid reactivity, TBARS; 4-hydroxyalkenals, 4HDA; sodium orthovanadate, SOV; Trigonella graecum seed powder, TSP; macrocyclic binuclear oxovanadium complex, MBOV; N(1)-2,4-dihydroxybenzylidene-N(4)-2-hydroxybenzylidene-S-methyl-thiosemicarbazidato-oxovanadium (IV), VOL; vanadyl sulphate, VSO4; metformin-decavanadate, MV10 [29][45][46][47][48][49][50][51][52][53][54][55][56][57][58][59][60][61][62][63][137,196,197,198,199,200,201,202,203,204,205,206,207,208,209,210,211,212,213,214].
The vanadium salt in oxidation state IV, vanadyl sulfate (VOSO4), has also been studied extensively. An early study showed that TBARS levels were elevated in vanadyl-treated animals, although cataract development was suppressed in STZ-diabetic Wistar rats [64][215]. By contrast, many reports showed in different tissues that the treatment with VOSO4 reversed the increased levels of LPO in response to diabetes induction [54][55][56][57][60][61][205,206,207,208,211,212] (Figure 1). These results were recently expanded to show similar normalization of the oxidative state in cardiac, lung, skeletal muscle and eye lens tissue [65][216]. Similar antioxidant effects were observed in the pancreas, liver and kidneys of diabetic rats treated with a macrocyclic binuclear oxovanadium complex (MBOV) [62][63][213,214] and in the pancreas and brain of diabetic rats treated with the N(1)-2,4-dihydroxybenzylidene-N(4)-2-hydroxybenzylidene-S-methyl-thiosemicarbazidato-oxovanadium (IV) (VOL) compound [58][59][209,210] (Figure 1).
Oxidative stress in the liver and muscle tissues of alloxan-induced diabetic rats was addressed after treatment with (H2Metf)3[V10O28] (metformin-decavanadate, MV10) [53][204]. After 60 days, decreased activity levels of SOD and CAT induced by alloxan were restored to normal levels (Figure 1). Furthermore, the increased levels of LPO markers in the diabetic animals were normalized after Metf-V10 treatment. This was observed for both MDA and 4-hydroxyalkenal (4HDA) levels in a similar fashion to treatment with insulin, while metformin alone had very limited effects [53][204] (Figure 1). Decavanadate was previously reported to increase the glucose uptake in rat adipocytes, in the presence or in the absence of insulin [66][217]. Together, these findings suggest that vanadium compounds are not only insulin mimetics but may also enhance the activity of insulin [4][67][68][23,164,218].
Changing the oxidation state of the vanadium compound changes the redox properties of the complex alters the formation of LPO products as described above for vanadate (VV) and VOSO4 (VIV). One study compared the effects of vanadium in oxidation states III, IV and V in a series of coordination complexes with the same ligand, chloro-substituted dipicolinic acid [52][203]. VIVdipic-Cl and VVdipic-Cl complexes in liver tissues produced improved blood glucose levels, while there were lesser effects of VIIIdipic-Cl [52][203] (Figure 1). This demonstrated that even high-oxidation-state vanadium compounds are beneficial in changing MDA levels toward normal and reducing ROS levels presumably through redox cycling. For complexes with the dipic-Cl ligand, it was surprising that the VV complex showed a trend towards being slightly better at normalizing the redox state of diabetic cells, though VV complexes would need to undergo Fenton chemistry first [52][203].
It is important to note that the animal models of diabetes using diabetogenic chemicals cause the destruction of β-cells resulting in type 1 diabetes, so it is unclear whether such effects would be observed in type 2 diabetes animal models or patients. Moreover, in some cases, the diabetic animals did not show a reduction in the activity of enzymes involved in antioxidant defense in all tissues analyzed. As an example, an increase in enzyme activity was observed in diabetic heart tissues when compared to normal animals [29][50][137,201], although even in such cases, treatment with vanadium compounds restored levels close to normal (non-diabetic) values [29][47][50][55][137,198,201,206].
Taken together, these data suggest treatment with vanadium compounds may contribute to alleviating oxidative stress in patients with diabetes and contribute to an overall improvement in metabolic function. However, more evidence of vanadium antioxidant beneficial effects and safety is still required.
3. Vanadium Lipid Peroxidation and Neurodegenerative Diseases
Vanadium is known to have neurotoxic effects and contribute to a number of neurodegenerative diseases presumably through the introduction of oxidative stress and LPO production. The brain contains high amounts of PUFAs, making it a prime target for LPO, which can cause the destruction of the myelin sheath, loss of neurons via cell death, disruption of the cell membrane potential, depletion of dopamine, and inactivation of phosphatase enzymes. Neurons are surrounded by a myelin sheath which is important for the development of the electric potential and the ability to transmit electrical impulses in the form of action potentials quickly. Vanadium exposure has been reported to cause damage to the myelin sheath [69][219] and, as a result of LPO, neuronal death. LPO in the mitochondria also leads to cell death through effects on mitochondrial membranes. Vanadium accumulates in the brain after exposure [70][220], indicating that the toxic effects of vanadium relating to membrane destruction may play a role in the reported neurodegenerative diseases such as Parkinson’s and Alzheimer’s. The metal content and transporters in the rat brain have been reported to be sensitive to the presence of other metals, including Mn, chromium, zinc, cobalt, aluminum, molybdenum and vanadium [71][221].
3.1. Parkinson’s Disease
Parkinson’s disease (PD) is a neurodegenerative disease that has been associated with several failures in brain function. A decrease in the neurotransmitter dopamine has been correlated with the onset of Parkinson’s which, with disease progression, leads to a failure in the dopaminergic system. Some basis of knowledge around metals, specifically manganese (Mn), and the onset of Parkinson’s or the onset of similar symptoms called Parkinsonism exists [72][222]. The latter is a condition that results in loss of motor and neurological function similar to that of Parkinson’s but does not exhibit the symptoms of Parkinson’s disease. Symptoms produced by Mn are called manganism [73][223]. Mn, like vanadium, undergoes redox cycling and is known to have many neurotoxic effects. The ability of Mn to produce ROS has been well characterized [74][224] and shown to cause effects on mitochondrial function similar to those observed with vanadium treatment, including the loss of the mitochondrial membrane potential and the release of Cyt C [75][225]. Additionally, LPO products have been observed in mitochondria and the endoplasmic reticulum system and are similar to the oxidative stress in response to manganese exposure. With high doses of Mn, symptoms of Parkinson’s disease are seen and correlated with the onset of Parkinson’s [76][226]. Given the similarities between vanadium and manganese, the effects of vanadium on the onset of Parkinson’s are likely to be similar. Ngwa and coworkers have reported a link between vanadium neurotoxicity and its effect on the dopaminergic system due to its effect on protein kinase C-delta and its function in cell signaling mechanisms [77][227]. Ohiomokhare and coworkers (2020) found that vanadium increased ROS and decreased motor function in Melanogaster drosophila, both wild-type and PD models, and that these effects were alleviated with chelators or the administration of L-DOPA [78][228].
3.2. Alzheimer’s Disease
Alzheimer’s disease (AD’s) is a neurodegenerative disease characterized by loss of memory. Although no single cause of AD’s has been discovered, there is evidence that metals, lipid peroxidation and oxidative stress can play a role in disease progression. The disease is associated with the accumulation of β-amyloid plaques in the brain that have the capability of interacting with redox-active metals, such as copper, zinc and iron [79][229]. These metal ions induce the disease because of their ability to generate ROS and damage the brain through DNA damage and oxidation of lipids and proteins. Studies have shown that 4-HNE, a product of LPO, is present in the brains of AD’s patients [80][230]. Mitochondrial ROS production and mitochondrial dysfunction have been associated with AD [81][231]. All of these are known products of vanadium-based oxidative stress and offer a basis for vanadium having a role in AD.
As is the case for Parkinson’s, there are only a limited number of studies characterizing vanadium effects on the development of AD’s disease or its progression. However, due to vanadium’s redox properties and ability to generate ROS, it has the potential to induce at least some similar effects. Montiel-Flores and coworkers found that the inhalation of vanadium pentoxide caused AD-like neuronal cell death in rats [82][232].
There is also a growing body of work investigating the use of vanadium in treating AD’s disease. Although vanadium has toxic effects, studies reported some potential of vanadium-based therapeutics for AD’s disease [83][233]. Vanadyl acetylacetonate was found to promote glucose and energy metabolism in neuronal cells but did not reduce β-amyloid plaque production [84][234]. Two peroxovanadium complexes were reported to inhibit β-amyloid fibril formation. He et al. (2015) showed that two complexes were able to inhibit the aggregation of amyloids using PrP106–126 and Aβ1–42, where PrP is from the prion disease and refers to protein-prion protein. Inhibition was more effective in PrP than in Aβ, but there was not much difference in its effects on cell toxicity. Peroxovanadium complexes increased cell viability perhaps due to the ability of peroxovanadium complexes to reduce methionine residues [85][235]. This group also found that BEOV was able to ameliorate AD symptoms through a number of mechanisms including inhibition of Aβ aggregate formation [86][236]. These results should encourage studies on the use of vanadium in the treatment of neurodegenerative diseases.
4. The Potential for Lipid Peroxidation as a Future Target for Therapeutic Treatments
4. The Potential for LPO as a Future Target for Therapeutic Treatments
The ability of vanadium compounds to impact oxidative stress and the formation of LPO products is well documented [87][237]. Since vanadium remains a comparatively underexplored metal [88][238], new compounds are being assayed to determine their potential for alleviating oxidative stress [89][90][239,240]. Novel compounds are being designed which affect LPO but lack cellular toxicity. New pathways are discovered by investigating organisms not traditionally investigated [91][241]. New approaches are being developed based on combatting oxidative stress in disease processes. For example, a 2D vanadium carbide synthetic enzyme referred to as V2C MXenzyme has been reported to alleviate ROS-mediated inflammation [74][224]. Specifically, the 2D V2C MXenzyme can replace SOD, CAT, POD, TPx, GPx and HPO, thus mimicking the intracellular antioxidant defense system against ROS-mediated oxidative damage including protein carbonylation, lipid peroxidation and DNA damage. In vitro and in vivo experiments demonstrated that V2C MXenzyme was biocompatible and exhibited ROS-scavenging capability, protecting cellular components against oxidative stress. Future investigations are likely to involve the characterization of novel biological systems, new compounds and agents such as the V2C MXenzyme and related systems [74][224] designed to combat the effects on oxidative stress and LPO.