2. LMNA-Related Dilated Cardiomyopathy
Dilated cardiomyopathy (DCM) is characterized by enlargement and dilatation of one or both ventricles of the heart, which occurs together with impaired contractility and heart function
[22]. The
LMNA gene is the second most commonly mutated gene in familial dilated cardiomyopathy (DCM), accounting for 5% to 8% of cases
[23]. Patients carrying pathogenic
LMNA mutations have a poor prognosis due to the high rate of sudden cardiac death resulting from malignant arrhythmias. Atrial fibrillation (AF), atrioventricular (AV) conduction block, ventricular tachycardia, and sudden cardiac death often precede the development of systolic dysfunction
[24][25][26][24,25,26]. Although
LMNA-related DCM is an adult-onset disease, it cannot be excluded that structural changes and arrhythmias may be present in early asymptomatic individuals
[27].
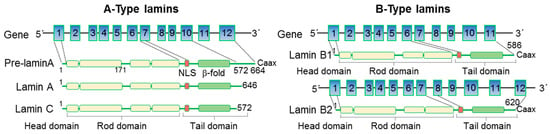
To date, around 500 mutations and 300 protein variants have been reported for
LMNA; detailed information on the different mutations is available through the UMD-
LMNA mutation database (
www.umd.be/LMNA, accessed on 3 January 2023). Most of the mutations associated with cardiomyopathies are located in the head and rod domains and are mostly truncation or missense mutations
[28]. Heterozygous truncation mutations often result in lamin A/C haploinsufficiency due to a premature termination codon generated by insertions or deletions resulting in a frameshift, aberrant splice site, or nonsense mutations. A homozygous
LMNA nonsense mutation (Y259X) has also been reported, resulting in a lethal phenotype
[29].
LMNA missense mutations, on the other hand, are thought to mostly act through a dominant negative mechanism
[28]. Patients carrying heterozygous mutations in
LMNA in combination with mutations within other genes such as
TTN, DES, SUN1/2, etc., display a particularly severe clinical cardiac phenotype
[30][31][32][33][34][30,31,32,33,34].
Another mutation often used for modeling the
LMNA LOF mutation is the p.R225X mutation, a nonsense mutation causing premature truncation of both lamin A and lamin C splice isoforms. Patients carrying this pathogenic mutation show early onset of AF, secondary AV block, and DCM
[35][87]. Like
Lmna−/− mice, homozygote
Lmna R225X mice also exhibit retarded postnatal growth, conduction disorders, and DCM
[36]. Other LOF mutations, e.g., K117fs and 28insA, also lead to a DCM phenotype.
LMNA p. K117fs mutation is a frameshift mutation that leads to a premature translation-termination codon
[37][38], whereas 28insA is an adenosine insertion mutation in exon 1 resulting similarly in a premature stop codon
[38][40]. Messenger RNAs (mRNAs) that contain a premature stop codon often undergo degradation through the nonsense-mediated mRNA decay (NMD) surveillance mechanism and thus can cause haploinsufficiency. Consistent with this, a significant decrease in lamin A/C protein levels is observed in K117fs iPSC-CMs as a result of NMD-mediated degradation of
LMNA mRNA
[37][38]. In addition to truncation mutations, which can result in
LMNA haploinsufficiency, mutations such as N195K, T10I, R541S, and R337H also show reduced lamin A/C protein levels
[39][40][35,41]. Patients carrying these pathogenic mutations also develop DCM
[26][41][42][26,51,88]. It is still unclear why these mutations lead to decreased lamin A/C levels. Possible reasons could be that protein translation or the stability of lamin A/C are affected in mutant CMs. For example, although
Lmna mRNA does not change, both lamin A and lamin C levels are decreased in CMs and MEFs derived from
Lmna N195K/N195K mutant mice
[39][35]. Interestingly, patients carrying different
LMNA missense mutations resulting in DCM also exhibit lower protein levels
[43][89]. To what extent the decrease in lamin A/C levels or changes in protein function result in disease pathogenesis is still largely unknown and needs further investigation.
Although it may seem that DCM is predominantly caused by
LMNA haploinsufficiency, missense mutations in
LMNA, which do not lead to changes in lamin A/C protein levels, also result in DCM. For example,
LMNA K219T missense mutation causing severe DCM and heart failure with conduction system disease
[44][52] does not lead to obvious changes in lamin A/C levels in K219T iPSC-CMs
[45][53].
LMNA H222P missense mutation has been shown to cause Emery–Dreifuss muscular dystrophy (EDMD) and DCM in patients. Homozygous mice with the H222P mutation display muscular dystrophy, left ventricular dilatation, and conduction defects and die by 9 months of age
[46][76]. Similarly to the K219T mutation, Western blot analysis of cardiac and skeletal muscle samples shows no obvious difference in lamin A/C protein levels between wild-type and
Lmna H222P/H222P mice
[47][74]. Interestingly, recent studies suggested a developmental origin of
LMNA-related cardiac laminopathy.
Lmna H222P/H222P embryonic hearts showed noncompaction, dilatation, and decreased heart function already at E13.5
[48][75], while
Lmna+/− and
Lmna−/− embryonic hearts showed noncompaction cardiomyopathy with no decrease in ejection fraction
[49][63]. Differentiation of mouse embryonic stem cells (ESCs) harboring the
Lmna p.H222P mutation revealed decreased expression of cardiac mesoderm marker genes, such as
Eomes and
Mesp1 as well as cardiac progenitor (CP) markers and impaired CM differentiation. This is in stark contrast to
Lmna+/− and
Lmna−/− mESCs, which showed premature CM differentiation
[48][49][63,75], suggesting different mechanisms behind the heart phenotype caused by lamin A/C haploinsufficiency or changes in protein functionality.
Among laminopathy-associated missense mutations, the addition of proline is the most common. Proline addition can significantly alter protein structure. For example,
LMNA S143P missense mutation causes DCM and disturbs the coiled-coil domain, thus affecting lamin A/C assembly into the nuclear lamina. This results in nuclear fragility and reduced cellular stress tolerance
[50][49]. The addition of proline might also affect protein phosphorylation through proline-directed kinases, such as the mitogen-activated protein (MAP) kinases, cyclin-dependent protein kinase 5 (CDK5), glycogen synthase 3, etc. Mutations resulting in the addition of proline often result in striated muscle disease, suggesting a common underlying mechanism
[51][90].
3. Arrhythmogenic Right Ventricular Cardiomyopathy
Arrhythmogenic right ventricular cardiomyopathy (ARVC) is an inherited heart muscle disorder that predominantly affects the right ventricle
[52][91]. A progressive loss of myocytes and fibro-fatty replacement associated with arrhythmias in the right ventricular myocardium is a hallmark of the disease
[53][92]. Mutations in desmosomal genes, such as Plakophilin 2 (
PKP2), Desmoplakin (
DSP), Desmoglein 2 (
DSG2), Desmocollin 2 (
DSC2), and junction plakoglobin (
JUP), are the main cause of ARVC
[54][55][56][57][58][59][93,94,95,96,97,98]. In addition, mutations in the calcium-handling protein Ryanodine Receptor 2 (
RYR2)
[60][99], Phospholamban (
PLN)
[61][100], the adherens junction protein Cadherin 2 (
CDH2)
[62][101], Integrin-Linked Kinase (
ILK)
[63][102], the signaling molecule Transforming Growth Factor-β3 (
TGFB3)
[64][103], the cytoskeletal structure protein Titin (
TTN)
[65][104], Desmin (
DES)
[66][105], transmembrane protein 43 (
TMEM43), and lamin A/C (
LMNA) have also been reported in ARVC
[24][67][68][69][24,106,107,108].
In 2011, Quarta et al. first reported ARVC caused by mutations in
LMNA. Four
LMNA variants were identified: R190W, R644C, R72C, and G382V
[24]. The R190W and R644C variants also cause DCM and left ventricular noncompaction (LVNC). In addition, R644C can also lead to lipodystrophy and atypical progeria, thus showing an extreme phenotypic diversity. ARVC patients with these four mutations all exhibit RV dilatation and systolic dysfunction. Histological examination of the right ventricular myocardium from R190W and G382V patients showed a loss of more than 50% of myocytes and extensive interstitial fibrosis and fatty replacement
[24]. Interestingly, immunohistochemical staining showed significantly reduced plakoglobin expression at the intercalated discs in the myocardium, which could contribute to the development of ARVC
[24]. M1K, W514X, and M384I mutations in
LMNA have also been identified in ARVC. Patients with M1K and W514X mutations show RV dilatation, non-sustained ventricular tachycardia, and complete atrioventricular block
[69][108]. A patient with the M384I variant not only developed ARVC but also peripheral neuropathy and peroneal muscular atrophy
[70][109].
So far, it remains unknown how
LMNA mutations result in ARVC. Since
LMNA is a ubiquitously expressed protein, its mechanoprotective function in cardiomyocytes, which can limit the progressive loss of myocytes, its role in the regulation of genes involved in cardiac contractility, and its important role in regulating cell fate choices, which may result in an excess of fibroblasts and adipocytes, might be involved. Tracing back the origins of fat tissue in a mouse model of ARVC, Lombardi et al. suggested that second heart field (SHF)-derived progenitor cells switch to an adipogenic fate through nuclear plakoglobin (JUP)-mediated Wnt signaling inhibition
[71][110]. A subset of resident cardiac fibro-adipocyte progenitor cells characterized by PDGFRA
posLin
negTHY1
negDDR2
neg expression signatures have been shown to be a major source of adipocytes in ARVC caused by Desmoplakin (
DSP) haploinsufficiency
[72][111]. Furthermore, the endocardium, epicardium, and cardiac mesenchymal stromal cells also serve as a source of adipocytes in the heart
[73][74][75][112,113,114]. Because the endocardium and epicardium give rise to diverse cardiac cell lineages, including mesenchyme and adipocytes
[76][115], via endothelial-to-mesenchymal transition (EndMT) and epithelial-to-mesenchymal transition (EMT), lamin A/C function in regulating EMT
[48][75] might also be a key mechanism driving ARVC pathogenesis.
4. Left Ventricle Noncompaction Cardiomyopathy
Left ventricular noncompaction (LVNC) cardiomyopathy is a rare congenital heart disease resulting from abnormal development of the endocardium and myocardium. Patients with LVNC exhibit a thin compact myocardium and excessive trabeculation and can eventually develop progressive cardiac dysfunction followed by heart failure. LVNC can manifest together with other cardiomyopathies and congenital heart disease
[77][116]. Studies have identified various genes associated with LVNC, such as
TTN,
MYH7,
TNNT2,
LDB3,
MYBPC3,
ACTC1,
DSP,
CASQ2,
RBM20, and the intermediate filaments
DES [78][117] and
LMNA [79][118], with the two most affected genes being
TTN and
LMNA [80][119]. The first reported
LMNA mutant variant causing LVNC is R190W, which is also associated with familial DCM and ARVC
[81][56]. Another pathogenic
LMNA variant causing LVNC is
LMNA R644C. R644C mutation carriers show an extreme phenotypic diversity, ranging from DCM and LVNC to lipodystrophy and atypical progeria
[82][59]. Parents and colleagues reported four family members with the
LMNA R644C mutation, three of whom developed left ventricular noncompaction cardiomyopathy with normal LV dimensions and function and without evidence of dysrhythmias
[83][60]. Other mutations such as
LMNA V74fs, R572C, and V445E have also been associated with LVNC. Patients with the V445E missense mutation are characterized by an arrhythmogenic form of LVNC, suggested to be due to dysfunctional SCN5A
[80][84][58,119].
How
LMNA mutations result in LVNC and the mechanisms underlying the high phenotypic diversity are largely unknown. Two recent studies demonstrated that
Lmna H222P/H222P as well as
Lmna−/− and
Lmna+/− embryonic hearts exhibit noncompaction, suggesting these mouse models as important tools to study the developmental origin and the mechanisms behind
LMNA-mediated noncompaction cardiomyopathy
[48][49][63,75]. Interestingly,
the our
esearchers' own research own study revealed that
Lmna LOF results in abnormal cell fate choices during cardiogenesis, i.e., promotes CM and represses endothelial cell fate. Since the crosstalk between CMs and endothelial cells is instrumental for proper cardiac development and myocardial compaction
[85][120], abnormal cardiovascular cell fate choices and dysfunctional endothelium might also contribute to LVNC. Thus, understanding the link between alternative cell fate choices, changes in cell behavior, and tissue-specific phenotypes caused by pathogenic
LMNA mutations would be an important question to address in further studies.
5. Restrictive Cardiomyopathy
Restrictive cardiomyopathy (RCM) is a rare cardiac disease characterized by increased myocardial stiffness resulting in impaired ventricular filling. Patients with RCM show enlarged atria and diastolic dysfunction, while systolic function and ventricular wall thicknesses are often normal until the later stages of the disease
[86][87][88][121,122,123]. Although most causes of RCM are acquired, several gene mutations have also been identified in patients with RCM
[86][87][88][89][121,122,123,124]. The most common mutated genes found in RCM are sarcomere-related genes such as
TTN [90][125],
TNNI3 [91][126],
MYH7 [92][127],
ACTC1 [93][128], etc. Mutations in non-sarcomere genes such as
DES [94][129],
TMEM87B [95][130],
FLNC [96][131], etc., have also been reported. Recently, Paller et al. reported a truncation mutation of
LMNA (c.835 delG:p.Glu279ArgfsX201) in an RCM patient who had a significant biatrial enlargement, atrial fibrillation, and skeletal muscle weakness. Both right and left ventricular size and function were normal, and histological analysis revealed cardiac hypertrophy and focal interstitial fibrosis in the endomyocardial tissue
[97][61]. How
Lmna mutations cause RCM is not known; a plausible mechanism could be the activation of profibrotic signaling, as discussed below.
6. Molecular Mechanisms Resulting in LMNA-Related Cardiomyopathy Pathogenesis
Since
LMNA-related cardiomyopathies caused by distinct point mutations show phenotypic diversity, the precise molecular mechanisms resulting in disease pathogenesis are also distinct and complex. Taking into account the variety of different functions of the nuclear lamina, three central mechanisms have been suggested to drive disease pathogenesis.
The “mechanical hypothesis” proposes that disruption of the nuclear lamina causes increased nuclear fragility and increased susceptibility to mechanical stress
[98][132]. This hypothesis is supported by observations that CMs from patients or mouse models with lamin A/C mutations exhibit nuclear rupture, DNA damage, and cell cycle arrest
[42][49][99][100][63,65,88,133]. Interestingly,
Lmna−/− non-CMs subjected to stretch show significantly increased DNA damage, further supporting the notion that the elevated cell death could be due to the inability of
Lmna−/− CMs to respond adequately to mechanical stress
[49][63]. Importantly, a recent study revealed that disrupting the LINC complex and thereby decoupling the nucleus/nucleoskeleton from the mechanical forces transduced by the cytoskeleton increases more than fivefold the lifespan of
LMNA-deficient mice
[101][134], pointing to therapeutic opportunities for patients carrying mutations resulting in nuclear fragility.
Myriad studies have demonstrated a role of lamins in regulating MAPK, TGF-β, Wnt–β-catenin, and Notch signaling cascades
[102][103][135,136] and suggested that altered signaling is a key driver of
LMNA-related dilated cardiomyopathy. For instance,
LMNA-related cardiomyopathy shows a significant increase in myocardial fibrosis which contributes to left ventricular dysfunction and heart failure
[24][39][104][105][24,35,137,138]. Profibrotic signaling, such as TGF-β, MAPK, and ERK signaling, is activated in
Lmna H222P/H222P mice, and the partial inhibition of ERK and JNK signaling before the onset of cardiomyopathy in
Lmna H222P/H222P mice significantly reduces cardiac fibrosis and prevents the development of left ventricle dilatation and decreased cardiac ejection fraction
[105][106][107][108][138,139,140,141]. Indeed, therapies targeting intracellular signaling alterations are being developed in a preclinical setting
[109][142].
Since nuclear lamins anchor chromatin at the nuclear periphery, the “chromatin hypothesis” suggests that chromatin alterations as a result of
LMNA haploinsufficiency or mutation result in abnormal gene expression programs responsible for the disease phenotype
[98][132]. In the last years, a number of studies using iPSC-CMs or mESC-CMs uncovered changes in chromatin architecture coupled to transcriptional changes in different ion channels such as
SCN5A,
CACNA1A/C/D,
HCN4,
SCN3b, and
SCN4b, as well as
Pdgfb pathway activation, which might explain the arrhythmogenic conduction defects in
LMNA patients
[37][45][49][110][38,53,63,143].
7. Advances in Therapeutic Strategies for LMNA-Related Cardiomyopathy
The clinical management of
LMNA-related DCM includes pharmacological treatment with ACE inhibitors and beta blockers and implantable cardiac defibrillators (ICDs)
[111][112][173,174]. Heart transplantation or ventricular assist devices may also be required for patients in the end stages of heart failure
[111][112][173,174]. The inhibition of mTOR, MAPK, and LSD1 significantly rescues the
LMNA-related DCM phenotype in mice
[48][105][113][75,138,175], and a novel and selective p38 MAPK inhibitor is now in a phase 3 clinical trial in
LMNA-related DCM
[114][176]. In addition, CRISPR/Cas9-based genome editing strategies have been used in
LMNA-caused Hutchinson–Gilford Progeria Syndrome (HGPS) and show promising results
[115][116][117][177,178,179]. By using guide RNAs (gRNAs) that target
LMNA exon 11 to specifically interfere with lamin A/progerin expression, both Santiago-Fernández et al. and Beyret et al. show a reduced progerin expression and improvement in the progeria phenotype in an HGPS mouse model
[115][116][177,178]. However, off-target effects, e.g., resulting from insertion and deletions during non-homologous end joining (NHEJ), are a major concern. To overcome these limitations, CRISPR/Cas9-mediated base pair editing systems have been used in HGPS mice
[117][179]. Base pair editing systems could modify the genome without the need of double-strand DNA breaks or donor DNA templates
[118][180]. Two classes of DNA base editors have been reported: cytosine base editors (CBEs), which convert C:G to T:A, and adenine base editors (ABEs) which convert A:T to G:C
[119][120][181,182]. Systemic injection of a single dose of dual AAV9 encoding ABE and sgRNA into an HGPS mouse model significantly extends the median lifespan of the mice, improves aortic health, and fully rescues VSMC counts as well as adventitial fibrosis
[117][179]. Despite the power of the base pair editing technology, a major limitation is the inability to edit the genome beyond four transition mutations. Prime editing represents a novel approach which is not only suitable for all transition and transversion mutations but also for small insertion and deletion mutations
[121][183]. Similar to base pair editing, prime editing does not require double-strand DNA breaks or donor DNA templates
[121][183] and could be used in the correction of genetic cardiomyopathies.