Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Gabriela Pastuch-Gawolek and Version 4 by Jessie Wu.
Sugars, which are a structural element during both the synthesis of the aforementioned glycoconjugates and the preparation of polysaccharide or glycopolymer carriers of anticancer drugs, are widely distributed in nature and also constitute one of the main classes of natural compounds found in living organisms, where they perform important functions in many physiological and pathological processes.
- prodrug
- glycopolymers
- receptors
1. Introduction
One of the possibilities to improve the effectiveness and safety of chemotherapeutic agents is the development of targeted therapies that will allow the drug to selectively interact with cancer cells. The use of targeted drugs makes it possible to deliver the biologically active substance precisely to the pathologically affected site, preventing the uptake of such a drug by healthy cells and, consequently, reducing systemic toxicity. In such a case, it is possible to reduce the therapeutic dose of the drug to achieve its sufficient intracellular concentration compared to the traditional applied drugs. Designed molecules typically target specific enzymes, receptor proteins, and signaling pathways [1][2][8,9]. Obtaining such compounds is possible by covalent coupling of a therapeutic agent with an appropriate ligand (e.g., sugar, peptide, vitamin, protein, or antibody) acting as a selective transporter. The compound created in this way should act selectively by targeting receptors on the membrane surface of diseased cells, to which the attached targeting ligand shows affinity and to which expression in the case of diseased cells is much higher than normal cells [3][10]. The success of such an approach depends to a large extent on the binding capacity of the ligand to the potential receptor, as well as the stability of the prodrug in the systemic circulation and its no-degradation before reaching target cells [4][5][11,12]. The structure of a targeted drug usually consists of several elements, such as a targeting ligand, a linker, and a specific active molecule (Figure 1). Each of these fragments should be meticulously designed because the effectiveness of the entire drug will depend on each of them. The targeting ligand plays a key role in the selective delivery of the drug to target cells that overexpress specific receptors. Its effectiveness is modulated by its specificity for tumor cells compared to normal cells. The linker between the ligand and the therapeutic payload should be designed to ensure the stability of the molecule in the systemic circulation and be easily cleaved upon reaching the target cells, thus releasing the attached drug responsible for exerting a specific pharmacological effect [3][4][5][10,11,12].
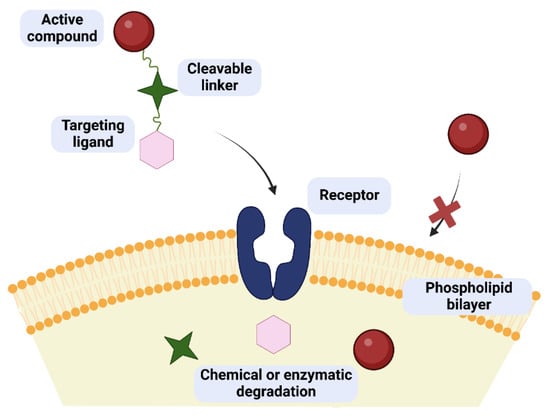
Figure 1. Concept of drug/glycoconjugate targeting in cancer therapy. The targeted drug can cross the cell membrane as opposed to the pure active compound. Inside the cell, the prodrug breaks down into its component parts.
2. Anticancer Drug Glycoconjugates
Despite significant advances in the diagnosis and treatment of cancer diseases, drug resistance still remains one of the main causes hampering the effectiveness of therapy. The emergence of resistance to therapy can occur at an early or later stage of treatment, thus limiting its success. It is important to understand the importance of altered glucose metabolism in driving cancer progression, response to treatment, and its role in resistance to commonly used drugs, such as doxorubicin, cisplatin, paclitaxel, and methotrexate, among others.
The first substance of natural origin used as an anticancer agent was podophyllotoxin (PDX). It is a potent anticancer agent but too toxic to be useful in the treatment of human neoplasms. Among many natural and synthetic derivatives of podophyllotoxin, two 4-demethylepipodophyllotoxin attached through O-glycosidic bond β-D-glucopyranoside cyclic acetals, known as etoposide (VP16-213) and teniposide (VM26), deserve attention. Teniposide is used in the treatment of childhood lymphoblastic leukemia, Hodgkin’s disease, non-Hodgkin lymphomas, and neuroblastoma. On the other hand, etoposide has activity against of testicular cancer, lymphomas, lung cancer, monocytic leukemia, non-Hodgkin lymphomas, and hepatocellular carcinoma [6][46].
The 1960s was a significant decade for the discovery of new anticancer drugs. During this period, Wani and Wallh isolated paclitaxel and the alkaloid, camptothecin. Camptothecin, obtained from extracts of CamptothecaCamptotheca acuminata Decne acuminata Decne (in 1873), showed good activity against L1210 leukemia, but, unfortunately, there are major limitations to its use as an anticancer agent, including toxicity, nonselectivity, and inactivation by human serum albumin (HSA). The search for improved analogs led to the discovery of topotecan (Hycamtin, N,N-dimethylaminomethyl substituent at the C-9 position of the parent structure) and irinotecan (Camptosar, prodrug of the 7-ethyl-10-hydroxycamptothecin analog), which were approved for clinical use [6][46].
One of the most effective and widely used anticancer drugs is paclitaxel (PTX, Taxol®), isolated from the bark of TaxusTaxus brevifolia brevifolia (Pacific Yew) and now also produced synthetically [7][47]. The first of several FDA approvals of various uses for Taxol® was announced in 1992 [8][48]. It is approved for the treatment of breast and ovarian cancers, AIDS-related Kaposi sarcoma, and a number of other cancers [7][47]. Although the initial response to paclitaxel is impressive, most breast cancer (BC) patients develop resistance, ultimately leading to relapse, metastasis, and death [9][49]. Unfortunately, paclitaxel has low oral bioavailability and very low selectivity [10][11][50,51]. The development of resistance of tumor cells and severe side effects in patients require further improvement of this drug.
An important class of chemotherapy drugs comprises anthracyclines, containing planar aromatic quinone rings connected to a sugar moiety. Doxorubicin (DOX), the active compound in the trade drug named adriamycin (ADM) and daunorubicin, which can be isolated from the bacterial strain Streptomyces peucetius, and epirubicin, idarubicin (semi-synthetic analog) belong to the most well-known antibiotics of the anthracycline family and are among the most prescribed drugs for the treatment of hematologic malignancies and adult solid tumors [12][52]. DOX, recognized by the World HealthWorld Health Organization Organization (WHO), shows efficacy against carcinomas of the breast, ovary, bladder, stomach, and thyroid, as well as small cell lung cancer, soft-tissue and osteogenous sarcoma, and numerous solid paediatric tumors. DOX demonstrates activity against hematopoietic malignancies, such as leukemias, lymphomas (Hodgkin and non-Hodgkin’s), and multiple myeloma. It is also the standard against which the cytotoxicity of new potential therapeutic agents is evaluated. Despite the therapy of DOX increasing survival in patients, it can also lead to many side effects, such as nephrotoxicity, hepatotoxicity, bone marrow suppression, irreversible cardiotoxicity, drug-induced leukemia, thrombocytopenia, anemia, liver toxicity, myelosuppression, vomiting, alopecia, and ulcerative stomatitis [13][53].
Since 1978, Pt-based antitumor drugs (cisplatin, carboplatin, and oxaliplatin) have been the first choice of chemotherapy drugs for malignant tumors, such as testicular, colorectal, non-small cell lung, ovarian, breast, head and neck, and nasopharyngeal cancer. However, their application is limited by severe side effects, such as renal toxicity, ototoxicity, neurotoxicity, and alopecia, as well as the problems of drug and cross-resistance and intrinsic or acquired resistance [14][54]. Oxaliplatin (Eloxatins®) is widely used to treat colorectal cancer in combination with 5-fluorouracil (5-FU) and leucovorin (LV). Unfortunately, oxaliplatin also causes many side effects, such as gastrointestinal toxicity, bone marrow suppression, and neuro- or nephrotoxicity. The low solubility in water and the slow excretion of oxaliplatin are the causes of the accumulation of metals [15][16][55,56].
Modern drug design often ensures selective and effective drug delivery to cancer cells with less toxicity to healthy cells. The unique glucose metabolism of cancer cells is the driving force behind the development of anticancer drug glycoconjugates (ADGs) designed for selective uptake by cancer cells. As a result of the high solubility in water, low toxicity, and high biocompatibility, the sugar moiety is an attractive system to facilitate drug delivery. Glycoconjugation is a very good method of imparting increased aqueous solubility to hydrophobic scaffolds, including several drugs such as aspirin [17][57], warfarin [18][58], and oxaliplatin [19][59].
So far, many semi-synthetic derivatives of anticancer drugs have been synthesized, and their effect on cytotoxicity has been investigated in terms of structure–activity relationships (SARs). During 2000 to 2021, fifty-four carbohydrate-based agents that contain sugar moieties as the major structural units have been approved as drugs or diagnostic agents, including three (Ambrucin, Japan, 2002; Mifamurtide, 2009, Europe, and Midostaurin, USA, 2017) with anticancer activity [20][60]. Glycosylation of therapeutic agents many times has been found to improve their pharmacokinetic parameters, reduce adverse effects, and expand half-life compared to the parent (not glycosylated agent). In this chapter, rwesearchers present the selected glycosylated therapeutic agents and the effect of attached sugar derivatives on the anticancer activity of those glycoconjugates.
The first glycoconjugate targeting GLUT1 transporters described in 1995 was glufosfamide, wherein β-D-glucose was connected to an alkylating agent, ifosfamide [21][22][61,62]. The task of the prodrug designed in this way was to increase the selectivity of ifosfamide (DNA alkylating drug) and reduction its toxicity [22][62]. In 1997, the first human clinical trials of glufosfamide were conducted in Europe [23][63]. A little later, research was continued in Japan and the USA, with promising results [24][64].
Then, various anticancer drug glycoconjugates targeted to GLUT or ASPGR were developed, based on cytotoxic molecules, such as chlorambucil (CLB) [25][65], azomycin [26][66], doxorubicin (adriamycin) [27][67], and paclitaxel [28][29][68,69]. Glycoconjugated prodrugs reported to date consist of a known anticancer drug directly linked via a glycosidic bond to a sugar unit or, as it is in most cases, a known anticancer drug joined to a sugar unit via linkers (esters, amides, ureas, and succinic acids, Figure 2). Commonly used sugar moieties include D-glucose, glucuronic acid, and D-galactose, as well as, to a lesser extent, L-rhamnose and D-xylose.
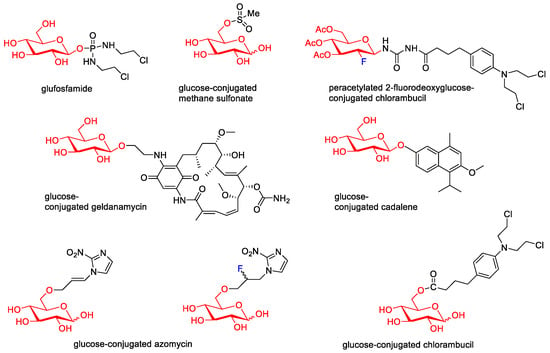
Figure 2.
Representative anticancer drug glycoconjugates.
Numerous troublesome side effects observed for doxorubicin therapy attracted the attention of scientists and contributed to the development of methods to increase its efficacy and, above all, reduce toxicity. Cao et al. designed the molecular hybrid of doxorubicin and 2-amino-2-deoxy-D-glucose, linked via a succinic linker. The 2DG-SA-DOX prodrug, targeting cancer cells by GLUT1, showed higher activity in cancer cell lines (MCF-7 and MDA-MB-231) and lower toxicity to normal cells than DOX, as well as lower organ toxicity [27][67].
Galactose residues could be specifically recognized by the asialoglycoprotein receptor (ASGPR), which is highly expressed in liver tissues. To explore the possibility of using galactosylated compounds in cancer-targeted therapy, doxorubicin was covalently conjugated with Gal to form a prodrug (Gal-DOX1). The antitumor efficacy of Gal-DOX1 in vitro was assessed in HepG2, MCF-7, and L02 cell lines. The cell viability of tested tumor cells incubated with DOX was higher than that of Gal-DOX1. At the highest dose of 10 μg/mL, the proliferation inhibition of the Gal-DOX1 was 54.3% of total cells, which is much higher than that of DOX (40.1%). Normal cells L02 with lower ASGPR1 level showed high cell viability, suggesting the low cytotoxicity of Gal-DOX1 in cells with low ASGPR1 low expression cells [30][70].
In 2018, a theranostic prodrug containing doxorubicin and a galactose moiety connected via a linker (see Figure 3) was used to target asialoglycoprotein receptors [31][71]. Activation of this prodrug (Gal-DOX2), by β-galactosidases in the colon, releases the parent drug and thus induces a fluorescence phenomenon that allows the monitoring of both the location and site of action of the drug. The imaging properties and therapeutic efficacy of Gal-DOX2 have been demonstrated both in vitro and in vivo in colorectal cancer models (HT-29 and HepG2). The cytotoxicity assays used showed that Gal-DOX2 exhibited a threefold more potent therapeutic effect in HT-29 cells than the HeLa cells. An additional advantage of this glycoconjugate with a simple structure, from a pharmacoeconomic point of view, is its ‘synthetic availability’. In comparison, doxorubicin conjugates with fatty acids, such as α-linolenic acid (LNA) and docosahexaenoic acid (DHA), generated by amide and ester linkages, and as single or double modifications (DOX-monoLNA, DOX-monoDHA, DOX-diLNA, DOX-diDHA) showed lower cytotoxicity against the tested cancer cell lines (SW480, SW620, and PC-3) compared to DOX but higher selectivity than DOX [32][72].
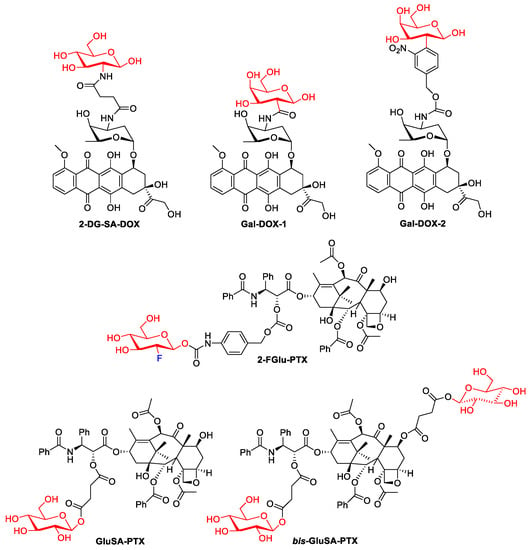
Figure 3.
Representative DOX glycoconjugates and PTX glycoconjugates.
In 2020, Meng et al. showed that the introduction of fluorodeoxyglucose (2F-Glu) or deoxyglucose in the paclitaxel skeleton significantly improves its solubility (PTX: 0.01 mg/mL, prodrug 2-FGlu-PTX: 0.48 mg/mL, Glu-PTX: 0.87 mg/mL). Glycoconjugate 2-FGlu-PTX exhibited sustained release with less than 50% hydrolysis detected after 12 h, while the non-fluorinated glucose conjugate was unstable (spontaneous release of paclitaxel was observed). 2F-Glu-paclitaxel showed increased cytotoxicity and selectivity (compared to PXL and Glu-PXL) to certain cancer cells (HepG2, NCI-H460, MCF-7) [33][73].
In another study, glycoconjugates of paclitaxel with single or double glucose moieties attached by succinate linkers were reported [34][74]. Both the C-2′-single glycosylated paclitaxel (GluSA-PTX) and the double glycosylated paclitaxel (bis-GluSA-PTX) conjugates showed effective cytotoxicity against breast cancer cells and improved hydro solubility compared to the parent drug. The improved solubility obtained is very important because it allows the elimination of the toxic surfactant CremophorCremophor EL EL (polyethoxylated castor oil), which has so far been used as a carrier in paclitaxel treatments.
The study presented by Han’s group showed that oxaliplatin modified with various sugar units, such as glucose, mannose, or galactose (Figure 4), has greater anticancer activity compared to the parent drug [35][75]. The tests suggest that the cytotoxicity of water-soluble platinum(II) complexes (Pt-1, Pt-2, Pt-3) is related to glucose transporters.
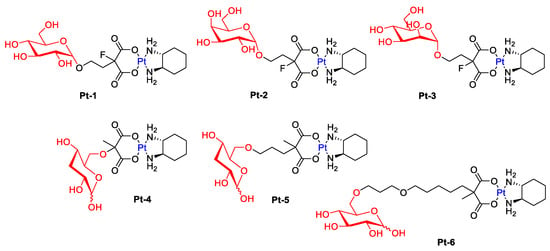
Figure 4.
Representative Pt-complex glycoconjugates (conjugated via C-1: Pt-1–Pt-3 and conjugated via C-6: Pt-4–Pt-6).
Patra et al. hypothesized that a modification at the C6 position of D-glucose should also have a positive effect on receptor binding (GLUT1) [36][76]. Furthermore, the Pt-6 complex (Figure 4) has been proven in vitro to preferentially kill cancer cells while exhibiting reduced accumulation and low toxicity to normal cells. The translocation efficiency and subsequent cellular accumulation decreased with increasing linker length.
An interesting example of glycoconjugated drugs is six glycosylated Pt(IV) compounds, presented in Figure 5, whose cytotoxicity was evaluated against five human cancer cell lines (Hela, HepG2, MCF-7, A549, and A549R) and normal human liver compared with cisplatin and oxaliplatin [37][77]. These complexes showed a higher level of apoptosis-inducing and lower cytotoxicity to normal LO2 cells than cisplatin and oxaliplatin while maintaining antitumor activity (Selectivity Index for Pt-10: 14.14, Pt-11: 24.1, Pt-12: 10.9, Cisplatin: 0.88, Oxaliplatin: 0.77). Additionally, the effect of alkyl ligands on cytotoxicity was greater than that of the ligand between the glycoligand and the Pt nucleus (Pt-9 had higher cytotoxic effects than Pt-10).
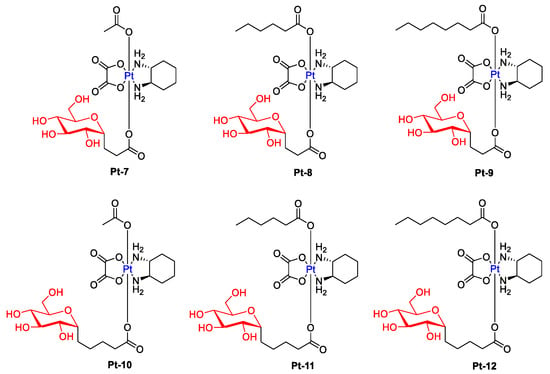
Figure 5.
Chemical structures of glycosylated Pt(IV) complexes with different linkers (Pt-7–Pt-12).
Vaidya and Patra presented an overview of glycoconjugation as an attractive strategy to impart selectivity and improve pharmacokinetics of platinum-based anticancer agents [38][78]. An interesting critical review article on the topic of progress (over the past decade 2010–2020) in glycoconjugation of anticancer drugs describes Fu et al. [39][79] and the Martin group [40][80]. The review works testify to the multitude of works carried out on improving the properties of platinum-based drugs using the glycoconjugation strategy and, consequently, the great interest in this subject. ResWearchers summarize selected examples of anticancer drug glycoconjugates (ADG) in Table 1.
Methotrexate (MTX, amethopterin), with both antiproliferative and anti-inflammatory properties, was introduced as a drug in 1953. It is used as a cytostatic drug in the treatment of cancers such as acute myeloid leukemia, acute lymphoblastic leukemia, breast, ovarian, lung, prostate, bladder, osteosarcomas, and solid tumors of the head and neck. It is also used in the treatment of autoimmune diseases such as psoriasis and rheumatoid arthritis [41][81]. MTX, as a folic acid antagonist, inhibits the activity of dihydrofolate reductase (DHFR), which catalyzes the conversion of dihydrofolate to tetrahydrofolate, which in turn is necessary for the synthesis of nucleotide bases. As a result, it leads to DNA and RNA synthesis disorders, inhibition of cell division, and, ultimately, cell death. The target of MTX action are all rapidly proliferating cells, including cancer cells, bone marrow, fetal cells, oral and intestinal mucosa, and bladder cells. MTX enters cells through protein folate transporters (RFC1 or FBP), and in higher concentrations, after saturation of transporters, through passive diffusion [42][82]. Although this medicine in the treatment of autoimmune diseases used in small doses is relatively safe, high doses used in oncological therapies cause very high systemic toxicity. Methotrexate, as a drug that acts nonspecifically on all body cells, can cause strong side effects, including, among others, gastrointestinal complaints, inflammation of the skin and blood vessels, kidney and liver failure, lung and intestinal diseases, and damage to the bone marrow and mucous membranes [43][83]. To avoid the undesirable consequences of the use of MTX, it is advisable to develop new strategies that will allow the selective delivery of methotrexate to the targeted sites, thus limiting side effects. Recently, many selective drug delivery systems have been developed [44][84]. MTX prodrugs activated under tumor microenvironment conditions were also obtained. An interesting example is glutathione-activated conjugate-based theranostic prodrug (Cy-SS-MTX) based on heptamethine cyanine (Cy) conjugated to MTX via a disulfide bond. This prodrug was activated by a high GSH level in the tumor, leading to a change in the optical properties of Cy group. This made it possible to track the activation of the administered prodrug under different excitation wavelengths. The obtained prodrug was tested both in vitro on four cell lines (MCF-7, SKOV-3, A549, and MCF-10A) and in vivo (mice bearing MCF-7 tumor). Based on the conducted research, it was found that prodrug improved anti-tumor efficiency of MTX and significantly reduced its toxicity to healthy cells [45][85]. However, there are few reports on the modification of the MTX molecule by its glycoconjugation. In 2001, the synthesis of conjugates was described, in which the carboxyl groups of the MTX molecule were connected to the anomeric position of per-O-acetylated-β-D-glucopyranosylamine through a linker made of lipoamino acids with three different alkyl chain lengths (LAAG-MTX). The cytotoxicity of the compound obtained against the lymphoblastic leukemia cell line (CCRF-CEM) was tested. Unfortunately, in vitro, this glycoconjugate turned out to be less active than the MTX derivative conjugated only with lipoamino acid [46][86].
Perhaps the failure of the first described MTX glycoconjugate made another MTX glycoconjugate described only in 2021 (Figure 6, MTX-Glu). It was designed on the assumption of the best affinity for GLUT receptors. Therefore, attached D-glucose molecules are not protected by protecting groups and have a β-configuration of the substituent at the anomeric carbon, as this orientation is preferred by GLUT proteins [47][87]. The 1,2,3-triazole ring in the linker between the sugar part and the drug increases the affinity of the compound for the transporter due to the possibility of hydrogen bonding. In turn, the linker structure with a glycosidic bond on the sugar side and a carbamate bond on the MTX side is selected to ensure the stability of the molecule in the extracellular environment and the possibility of chemoenzymatic degradation inside the cell [48][88]. The results of activity assays of the obtained MTX-Glu glycoconjugate allowed reusearchers to conclude that it has a strong cytotoxic effect in the in vitro environment against a wide panel of cancer cell lines, similar to the activity of unmodified MTX. This has also been confirmed in in vivo studies targeting breast cancer in mice. At the same time, the MTX conjugate showed low toxicity to noncancer cells, which significantly improved the selectivity of the drug. Additionally, the uptake of glycoconjugate by tumor cells and its accumulation in the intracellular compartment are significantly more efficient compared to MTX, which may indicate facilitated transport of the glycoconjugate by targeting GLUT1 transporters, its cellular distribution and intracellular release of the active molecule [49][50].
Table 1. Representative anticancer drug glycoconjugates.
Representative anticancer drug glycoconjugates.
| Drug | Conjugated Sugar | Type of Anticancer Activity Studies; Transportation Mode |
Activity Compared to Glycone/Properties | Ref. |
|---|---|---|---|---|
| Ifosfamide | D-Glucose | Alkylating agent | Glufosfamide
|
[22][62] |
| Doxorubicin (DOX, ADM) |
2-amino-2-deoxy-D-glucose and succinic acid |
Antitumor antibiotic GLUTs mediated |
2DG–SA–DOX
|
[27][67] |
| Doxorubicin (DOX, ADM) |
Galactose | Antitumor antibiotic | Gal-DOX1
|
[30][70] |
| Doxorubicin (DOX, ADM) |
Galactose | ASPG mediated | Gal-DOX2
|
[31][71] |
| Chlorambucil (CLB) | Amino derivatives of glucose, mannose, galactose, xylose, lyxose, D-threoside | Alkylating and DNA-complexing agent |
D-threoside-CLB
|
[52][92] |
| Chlorambucil (CLB) | Peracetylated 2-fluorodeoxyglucose | FDG-CLB
|
[53][93] | |
| Paclitaxel (PTX) | Glucose | Mitotic inhibitor | Glu-PTX
|
[28][29][54][68,69,94] |
| Paclitaxel (PTX) | Glucose | 2FGlu-PTX/PTX
|
[33][73] | |
| Paclitaxel (PTX) | Glucose | a single (GluSA-PTX) and double (bis-GluSA-PTX) bis
|
[34][74] | |
| Azomycin | Glucose | GLUTs-mediated | Glucoazomycins
|
[26][66] |
| Geldanamycin (GA) | Glucose | HSP90 inhibitor | Glu-GA
|
[55][95] |
| Geldanamycin (GA) | Galactose Lactose |
HSP90 inhibitor | Gal-GA and Lac-GA
|
[55][95] |
| Emodin (EM) | D-rhamnose | Tyrosine kinase inhibitor | Rha-EM
|
[56][96] |
| Platinum | Glucose | GLUTs mediated | Glucose-conjugated Pt(IV) complexes
|
[37][77] |
| Oxaliplatin | Glucose, Mannose Galactose |
GLUTs mediated |
|
[35][75] |
aASGP-R: asialoglycoprotein receptor (ASGP-R), GLUT1: glucose transporter 1, GLUT5: glucose transporter 5, PIC: polyion complex.
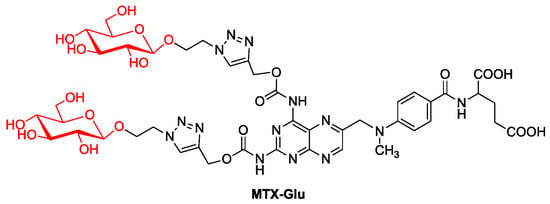
Figure 6. MTX glycoconjugate.
MTX glycoconjugate.