Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Lindsay Dong and Version 1 by Alessandra Torina.
Chagas disease is a chronic systemic infection transmitted by Trypanosoma cruzi. Its life cycle consists of different stages in vector insects and host mammals. Trypanosoma cruzi strains cause different clinical manifestations of Chagas disease alongside geographic differences in morbidity and mortality. Natural killer cells provide the cytokine interferon-gamma in the initial phases of T. cruzi infection. Phagocytes secrete cytokines that promote inflammation and activation of other cells involved in defence. Dendritic cells, monocytes and macrophages modulate the adaptive immune response, and B lymphocytes activate an effective humoral immune response to T. cruzi.
- Trypanosoma cruzi
- immunity
- toll-like receptors
- virulence factors
1. Introduction
The infection by Trypanosoma cruzi is responsible for a chronic and systemic disease known as Chagas disease that is recognized as a neglected disease by the World Health Organization. The parasite can infect humans and a lot of different species of wild and domestic animals and is mainly transmitted by bloodsucking reduviid insects of the Triatominae subfamily through three overlapping cycles: domestic, peridomestic and wild [1]. Triatoma infestans, Rhodnius prolixus and Triatoma dimidiata are the only competent vectors able to transmit T. cruzi to humans. Triatoma infestans is mainly spread in the sub-Amazonian endemic regions, R. prolixus is reported in South and Central America and T. dimidiata is reported in Mexico [2,3][2][3].
Other transmission routes to humans involve blood transfusion and vertical transmission [4,5][4][5]. In rare cases, ingestion of contaminated foods or liquids or raw meat with a massive parasite infestation can represent an additional transmission route [6]. The life cycle of T. cruzi includes several stages in vector insects and host mammals. In insects, the parasite assumes two typical forms identified as replicative epimastigotes and metacyclic tripomastigotes. In mammals, the typical forms are non-replicative blood tripomastigotes and replicative intracellular mastigotes [7]. Various T. cruzi strains circulate in mammalian hosts and in insect vectors. This heterogeneity can be responsible for the different clinical manifestations of Chagas disease as well as the differences in morbidity and mortality reported in different geographic areas [8,9][8][9].
In humans, Chagas disease usually evolves through an acute phase that can last up to two months, followed by an asymptomatic phase, also called an intermediate or indeterminate phase, and finally, a chronic phase. In most cases, patients affected by T. cruzi are asymptomatic or show mild symptoms. However, 10–30% of infected individuals show non-specific symptoms after an incubation period of 5–40 days. These symptoms include abdominal pain; anorexia; fever; malaise; lymphadenopathy; enlarged liver, spleen and lymphnodes; and localized or generalized subcutaneous oedema. In the case of vector transmission, T. cruzi inoculation can lead to the appearance of two typical clinical signs at the portal of entry [10]. One of them is the chagoma, a skin rash and oedema at the inoculation site, persisting for several weeks. The other is the Romaña sign which occurs after the accidental deposition of contaminated stools in the conjunctival sac due to eye rubbing. The transfer of tripomastigotes to the conjunctiva causes eyelid edema and conjunctivitis, which is often also associated with lymphadenitis or cellulitis. During the weeks following the bite, some patients may also develop a diffuse morbilliform rash [11,12][11][12]. In the case of oral transmission, patients may show nausea and vomiting, diarrhea, jaundice, abdominal pain and gastrointestinal bleeding. In 5–10% of cases, patients with an acute infection can die of myocarditis, encephalitis or meningoencephalitis, with most deaths usually occuring in children [13].
The pathogenesis of Chagas disease is not well understood. A long period of parasite persistence can lead to both direct and indirect injures. Direct injuries consist of T. cruzi-mediated cellular and neuronal damage, while indirect injuries are caused by the immune response and autoantigens [18,19][14][15].
The course of the disease is determined by the balance between the immune response, the inflammatory response of the host tissues and the replicative activity of the parasites [4,20][4][16]. Therefore, an ineffective immunological response will increase the size and persistence of a parasitic load and consequently lead to an excessive inflammatory response that causes tissue damage.
2. The Innate Immune Response to Trypanosoma cruzi
Innate immunity, consisting of phagocytes, especially macrophages, neutrophils and dendritic cells, constitutes the first line of defence that T. cruzi faces when invading a vertebrate host [21,22,23][17][18][19]. The role of these cells is to recognize pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) by membrane receptors such as toll-like receptors (TLRs). Trypanosoma cruzi can be recognized by TLRs and the cytokine production that is subsequently activated by these receptors has an important role in the host’s defence. Nucleotide-binding oligomerization domain-like receptors (NLRs) can also recognize T. cruzi; these receptors recognize PAMPs, which can be phagocytosed or can enter into cells through pores, and DAMPs [24][20]. Furthermore, phagocytes secrete cytokines involved in the promotion of inflammation and in the activation of other defence cells at the infection site. Trypanosoma cruzi antigens act as recognition signals and regulate the expression of proinflammatory cytokines from macrophages such as IL-1, IL-12, TNF-α and IL-10. Following T. cruzi infection, proinflammatory cytokines and IFN-γ produced by natural killer (NK) cells, together with effector T cells, influence macrophage activation status [25][21]. IFN-γ can activate macrophages through the classical pathway, even in combination with TNF-α. Another important component of innate immunity is the complement cascade system, which consists of several plasma proteins that are capable of opsonizing pathogens, thereby recruiting phagocytic cells to the infection point and destroying the infectious agent [23][19]. The complement cascade functions as a proteolytic enzyme cascade that amplifies signals generated by the pathogen infection and determines parasite elimination. Different pathways (classical, alternative and lectin) can lead to complement cascade activation, converging towards the cleavage of C3 into C3a and C3b [35][22]. C3a has a proinflammatory action, while C3b is recognized by neutrophil and macrophage receptors that promote pathogen phagocytosis. The inflammasome is a multimeric protein complex, which is assembled in the cytoplasm of host cells (which are also cells of innate immunity) following various types of stress signals or in presence of microbial molecules [36][23]. [36][23]. The inflammasome generally consists of three components: inflammatory caspases such as caspase-1, an adapter molecule such as ASC, and a sensor protein such as NLRP3.3. The Adaptative Immune Response to Trypanosoma cruzi
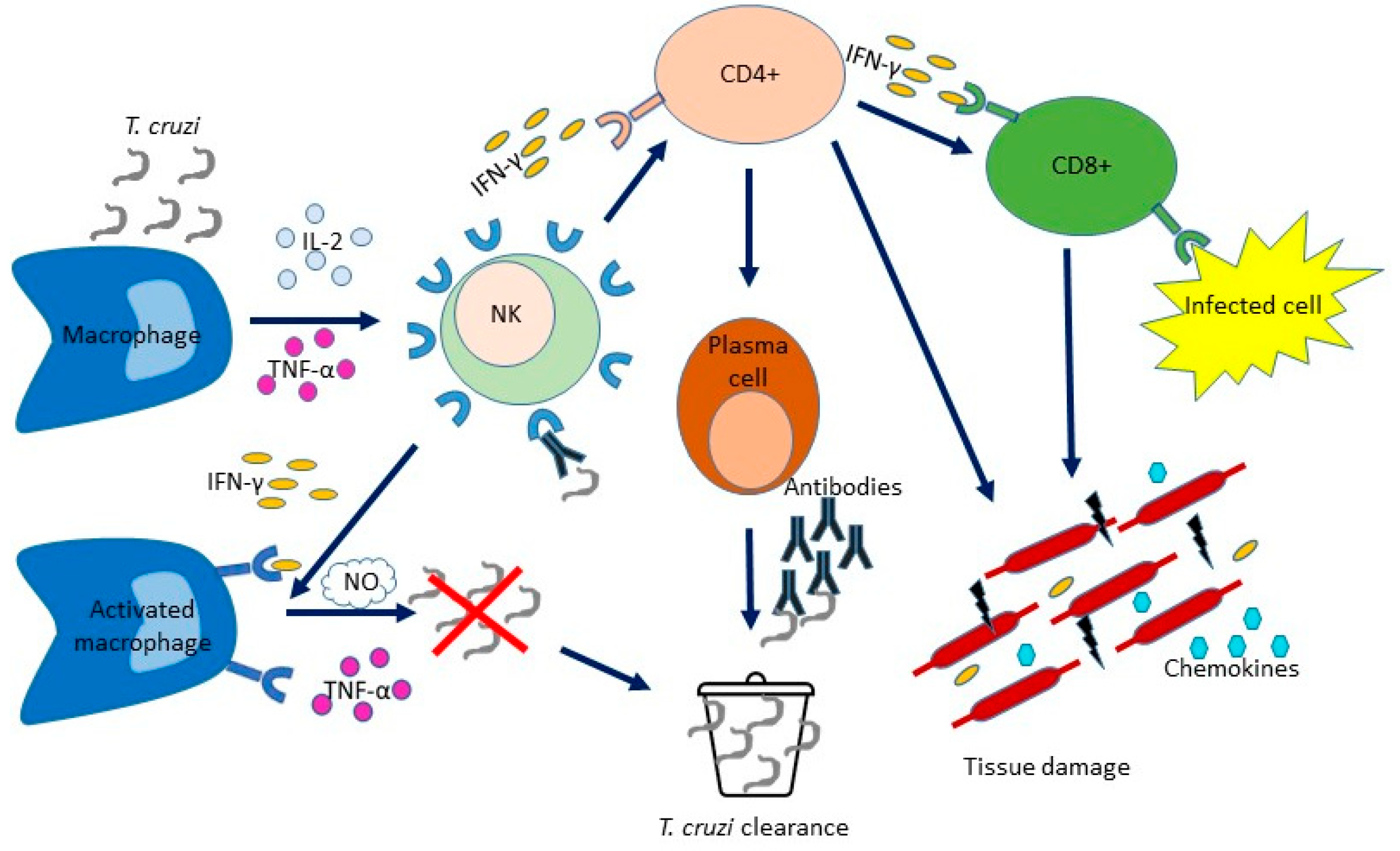
The onset of adaptive immunity is followed by the enhancement of circulating activated B lymphocytes that produce and secrete antibodies which play a crucial role in the adaptive humoral immune response. Kumar and Tarleton demonstrated that mice unable to produce antibodies could not control T. cruzi growth and died during the acute phase of the disease, thereby demonstrating the importance of the humoral immune response in controlling T. cruzi infection [37][24]. Although B lymphocytes mount an effective immune response to T. cruzi during its early stages [20,38][16][25], antibodies mostly produced against T. cruzi surface antigens may not completely resolve the infection and allow the parasite to permanently infect the host [23][19]. Cytokines largely coordinate both humoral and cellular immune responses to T. cruzi infection. B cells are fundamental in activating Th1 cell activities which favor the control of parasite growth [20][16]. A reduction of proinflammatory cytokines (IFN-γ, IL-12) has been shown in spleen supernatants from mice lacking mature B cells [20][16]; the immune system is unable to differentiate effector CD8+ T cells and to educate a Th1 functional model of T cell cytokines in the absence of mature B cells. T lymphocytes are crucial in the adaptive cellular immune response. Following the recognition of signals from T cell receptors on the surface of antigen-presenting cells (APCs), the T cell response is activated and naive T (Tn) cells undergo clonal expansion and change the molecular expression and cytokine production, thereby generating T cells with different roles [23][19]. The T cell activation process results in the production of effector T cells (TE) and also generates memory T cells that are capable of self-renewal and long-term persistence. The differentiation and expansion polarized towards IFN-γ of CD4+ and CD8+ T cells are induced by IL-12, which is produced by DC and NK cells, and trigger CD8+ T cells cytotoxic activity and macrophage effector mechanisms. CD4+ TE lymphocytes stimulate the proliferation of B lymphocytes and the production of antibodies that can determine the lysis of trypomastigotes. Moreover, in the acute phase of the infection, T cells are recruited to the tissues where IFN-γ induces the production of chemokines. A proper balance between inflammatory and anti-inflammatory cytokines and an adequate cellular response must be achieved to avoid tissue damage and keep parasite levels in check [39][26] (Figure 21).Figure 21. Immune mechanisms acting during Trypanosoma cruzi infection. T. cruzi infects nucleated cells. The first defence weapon against parasitic infection is mediated by the cells involved in innate immunity (macrophages, DC and NK cells), which occurs before the immune response by specific T and B cells. The production of IL-12 by macrophages is stimulated by T. cruzi antigens. IL-12 stimulates NK cells to produce IFN-γ. This, along with TNF-α activity, determines macrophage activation, induces the inflammatory process and controls pathogen replication. The levels of NO produced by macrophages correlate with the control of the parasite load. IL-12 produced by DC and NK cells stimulates the expansion of CD4+ and CD8+ T cells with polarization towards IFN-γ, thereby triggering the cytotoxic activities of CD8+ T cells cytotoxic and the effect activities of macrophages effector. B lymphocytes are stimulated to proliferate and produce antibodies by CD4+ T lymphocytes. During the acute phase, T cells are recruited to the tissues where IFN-γ induces chemokine production. Although the inflammatory environment is critical to the host’s resistance, it can also cause tissue damage.
4. Toll-like Receptors
Toll-like receptors (TLRs) are a family of pattern recognition receptors, which are shared by macrophages and other cells involved in innate immunity. TLRs act in the first stages of the immune response by recognizing different microbial structures/patterns [43,44,45][27][28][29].
Specific biological responses are elicited by TLRs via Toll/interleukin-1 (IL-1) receptor (TIR) domain-containing adaptor molecules, including MyD88, TRIF, TIRAP (Mal) and TRAM [46][30].
These receptors are involved in T. cruzi elimination and in phagocyte recruitment at the infection site [47,48,49][31][32][33]. However, the inappropriate activation of these receptors may be related to the establishment of a pathological condition [50,51][34][35].
Multiple TLR ligands of T. cruzi are able to activate the innate immune system response and, subsequently, the adaptative immunity response. The latter has been related to protection from the infection but also to pathogenesis [53][36]. In particular, it was reported that TLR2 activates the small guanine phosphonucleotide-binding protein Rab-5, which induces T. cruzi internalization by macrophages [54][37]. Moreover, when stimulated before the infection, TLR2 is able to promote the survival of infected mice [46,52][30][38]. TLR2 and TLR4 are able to sense glycoinositolphospholipids (GIPLs) and GPI anchors present on the trypomastigote cell surface [30[22][23][24][25][26][27][28][29][30][31][32][33][34][35][36][37][38][39][40][41][42][43][44][45][46][47][48][49],31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57], thereby inducing the activation of mitogen-activated protein kinase (MAPK) cascade and nuclear factor-kappa B (NF-κB) pathways. The result is NO and pro-inflammatory cytokine production and the activation of a Th1-type response [58][50].
Human neutrophils stimulated by T. cruzi generate neutrophil extracellular traps (NETs), which are fibrous traps of DNA, histones, and granules that are involved in pathogen killing. A study reported that NET release was reduced as a result of treatment with antibodies against TLR2 and TLR4 [62][51].
T. cruzi-infected macrophages are able to produce higher amounts of extracellular vesicles (EVs) with respect to non-infected cells. These EVs interact with TLR2 and induce translocation of NF-κB to the nucleus, thereby activating the production of pro-inflammatory cytokines (TNF-α, IL-6 and IL-1β) which are able to maintain the inflammatory response [63][52].
TLR2 inhibition increases the histopathological damage induced by parasites, reduces IL-6 and IL-10 secretion and the expression of proliferation and differentiation markers, while increasing the expression of cell death markers [68][53].
Galectin-3, a β-galactoside-binding lectin, acting in several biological processes [69][54], allow macrophages and epithelial cells to bind galactosides of membrane debris obtained from the vacuoles that are used to evade the phagolysosomal pathway of the host by some intra-vacuolar pathogens [45][29]. Galectin-3 is able to favor cellular infiltration in the hearts of mice infected by the pathogen, collagen deposition and cardiac fibrosis.
5. Virulence Factors
Trypanosoma cruzi, during its different stages, can infect different host cells using several virulence mechanisms: resistance to oxidative damage, humoral immune response evasion and cell invasion [71][55]. Different virulence factors act in a sequential manner during the different phases of the T. cruzi life cycle. Upon infection, metacyclic trypomastigotes (MT) mainly invade local macrophages, fibroblasts and tissues at the site of infection [72][56]. The antioxidant mechanisms used by T. cruzi are crucial for the inactivation of reactive oxygen and nitrogen species released by the host cells at the early stage of the infection [73][57]. The parasite produces several enzymes, such as peroxidases, that act on different molecules from the cellular oxidative pathway. Glutathione peroxidase TcGPXI (present in the cytosol) deactivates exogenous hydroper-oxides and TcGPXII (present in the endoplasmatic reticulum) inactivates lipid-hydroperoxides [74][58]. Ascorbate-dependent heme peroxidase TcAPX disables the binding of hydroxyl ions with oxygen in conjunction with the cytosolic tryparedoxin peroxidase TcCPX and mitochondrial TcMPX. T. cruzi also has iron superoxide dismutases (FeSOD) that detoxify reactive oxygen species generated in the cytosol, glycosomes and mitochondria [75][59]. The expression of enzymes of the T. cruzi antioxidative network is related with its life cycle. After transforming into bloodstream trypomastigotes (BT), T. cruzi is able to resist the humoral immune response and the lytic effects of the complement system [76][60]. The evasion mechanism is mediated by the surface glycoproteins of T. cruzi trypomastigotes, which restrict the activation of the classical and alternative complement pathways [77][61]. The trypomastigote decay-accelerating factor (T-DAF) is a surface glycoprotein that interferes with the C3 convertase-mediated assembly of the classical and alternative pathways [78][62]. The complement regulatory protein (CRP) is a surface-anchored glycoprotein expressed only by trypomastigotes, which inhibits the activation pathway of the complement system [77][61]. T-DAF and CRP are trans-sialidase-like glycoproteins belonging to the T. cruzi trans-sialidase superfamily [79][63]. Both proteins impair C3b formation by interacting with C4b and C3b [35][22]. Calreticulin (TcCRT) is a surface molecule that interacts with C1q to inhibit the activation of the classical complement pathway [80][64]. The complement C2 receptor inhibitor trispanning (TcCRIT) factor impairs the activation of complement cascades via both the classical and lectin pathways through the cleavage of the shared C2 factor and impairs the formation of C3 convertase via its interaction with C4 [35][22]. The proline racemases (PRs) TcPRACA and TcPRACB are secreted and intracellular enzymes, respectively [81][65]. TcPRACA is a B cell mitogen which initiates the activation of nonspecific polyclonal lymphocytes and is important for T. cruzi evasion and persistence [82][66]. The overexpression of TcPRAC isoforms results in increased parasite differentiation and cell invasion [83][67]. Tc52 is a secreted protein responsible for suppressing T cell proliferation [84][68]. It is able to modulate the expression of macrophage cytokines and iNOS and the production of NO [85][69]. Once they parasites have differentiated into extracellular amastigotes (EA), they start a new cycle of infection and invade new host cells, therefore they require molecules that allow them an efficient cellular invasion that favors adhesion and the activation of signaling cascades [86][70]. P21 and TcMVK proteins released by EAs favour host cell invasion. P21 rearranges actin filaments of the host cells and induces actin polymerization and phagocytosis [87][71]. TcMVK is bound to the membrane of the host cells and induces parasite uptake into HeLa cells [88][72]. T. cruzi has developed surface proteins (transialidases, mucins, mucin-associated surface glycoproteins and phospholipases) that allow the adhesion of metacyclic trypomastigotes and extracellular amastigotes to host cells through interactions with carbohydrates [86][70]. Gp82 is a surface protein of the metacyclic phase of T. cruzi that is responsible for adhesion to the host cell and activation of the Ca2+ signaling cascade, leading to internalization of the parasite [89][73]. Transialidase enzymes (TS) are important for T. cruzi virulence [90][74] as they allow the pathogen to acquire sialic acid from host cells and modify trypomastigote surface proteins, making them capable of inducing cell paralysis and cell lysis.References
- Deane, L.M. Animal reservoirs of Trypanosoma cruzi in Brazil. Rev. Bras. Malariol. Doenças Trop. 1964, 16, 27–48.
- Lent, H.; Wygodzinsky, P. Revision of the Triatominae (Hemiptera Reduviidae), and their significance as vector of Chagas disease. Bull. Am. Mus. Nat. History 1979, 163, 123–520.
- Galvão, C.; Carcavallo, R.; Rocha, D.S.; Jurberg, J. A checklist of the current valid species of the subfamily Triatominae Jeannel; 1919 (Hemiptera; Reduviidae) and their geographical distribution; with nomenclatural and taxonomic notes. Zootaxa 2003, 202, 1–36.
- Schmunis, G.A. Prevention of transfusional Trypanosoma cruzi infection in Latin America. Memórias Inst. Oswaldo Cruz 1999, 94 (Suppl. 1), 93–101.
- Bern, C.; Montgomery, S.P.; Katz, L.; Caglioti, S.; Stramer, S.L. Chagas disease and the US blood supply. Curr. Opin. Infect. Dis. 2008, 21, 476–482.
- Pereira, K.S.; Schmidt, F.L.; Guaraldo, A.M.; Franco, R.M.; Dias, V.L.; Passos, L.A. Chagas disease as a foodborne illness. J. Food Prot. 2009, 72, 441–446.
- Tyler, K.M.; Engman, D.M. The life-cycle of Trypanosoma cruzi. In American Trypanosomiasis; Tyler, K.M., Miles, M.A., Eds.; World class parasites; Kluwer Academic Publishers: Boston, MA, USA, 2003; Volume 7, pp. 1–11.
- Macedo, A.M.; Machado, C.R.; Oliveira, R.P.; Pena, S.D. Trypanosoma cruzi: Genetic structure of populations and relevance of genetic variability to the pathogenesis of Chagas disease. Memórias Inst. Oswaldo Cruz 2004, 99, 1–12.
- Manoel-Caetano, F.S.; Silva, A.E. Implications of genetic variability of Trypanosoma cruzi for the pathogenesis of Chagas disease. Cad. Saúde Pública 2007, 23, 2263–2274.
- Nobrega, A.A.; Garcia, M.H.; Tatto, E.; Obara, M.T.; Costa, E.; Sobel, J.; Araujo, W.N. Oral transmission of Chagas disease by consumption of acai palm fruit; Brazil. Emerg. Infect. Dis. 2009, 15, 653–655.
- Patel, S.; Sethi, A. Imported tropical diseases. Dermatol. Ther. 2009, 22, 538–549.
- Lupi, O.; Bartlett, B.L.; Haugen, R.N.; Dy, L.C.; Sethi, A.; Klaus, S.N.; Machado Pinto, J.; Bravo, F.; Tyring, S.K. Tropical dermatology: Tropical diseases caused by protozoa. J. Am. Acad. Dermatol. 2009, 60, 897–925.
- WHO. Control of Chagas Disease; Second report of the WHO Expert Committee; Technical report series no 905; World Health Organization: Geneva, Switzerland, 2002.
- Bonney, K.M.; Engman, D.M. Chagas heart disease pathogenesis: One mechanism or many? Curr. Mol. Med. 2008, 8, 510–518.
- Nagajyothi, F.; Machado, F.S.; Burleigh, B.A.; Jelicks, L.A.; Scherer, P.E.; Mukherjee, S.; Lisanti, M.P.; Weiss, L.M.; Garg, N.J.; Tanowitz, H.B. Mechanisms of Trypanosoma cruzi persistence in Chagas disease. Cell. Microbiol. 2012, 14, 634–643.
- Cardillo, F.; Postol, E.; Nihei, J.; Aroeira, L.S.; Nomizo, A.; Mengel, J. B cells modulate T cells so as to favour T helper type 1 and CD8+ T-cell responses in the acute phase of Trypanosoma cruzi infection. Immunology 2007, 122, 584–595.
- Rezende-Oliveira, K.; Sarmento, R.R.; Rodrigues, V., Jr. Production of cytokine and chemokines by human mononuclear cells and whole blood cells after infection with Trypanosoma cruzi. Rev. Soc. Bras. Med. Trop. 2012, 45, 45–50.
- Pinho, R.T.; da Silva, W.S.; de Castro Cortes, L.M.; da Silva Vasconcelos Sousa, P.; de Araujo Soares, R.O.; Alves, C.R. Production of MMP-9 and inflammatory cytokines by Trypanosoma cruzi infected macrophages. Exp. Parasitol. 2014, 147, 72–80.
- Acevedo, G.R.; Girard, M.C.; Gómez, K.A. The unsolved jigsaw puzzle of the immune response in Chagas disease. Front. Immunol. 2018, 9, 1929.
- Gurung, P.; Kanneganti, T.D. Immune responses against protozoan parasites: A focus on the emerging role of Nod-like receptors. Cell. Mol. Life Sci. 2016, 73, 3035–3051.
- Noel, W.; Raes, G.; Hassanzadeh Ghass, G.; De Baetselier, P.; Beschin, A. Alternatively activated macrophages during parasite infections. Trends. Parasitol. 2004, 20, 126–133.
- Lidani, K.C.F.; Bavia, L.; Ambrosio, A.R.; de Messias-Reason, I.J. The complement system: A prey of Trypanosoma cruzi. Front. Microbiol. 2017, 8, 607.
- Zamboni, D.S.; Lima-Junior, D.S. Inflammasomes in host response to protozoan parasites. Immunol. Rev. 2015, 265, 156–171.
- Kumar, S.; Tarleton, R.L. The relative contribution of antibody production and CD8+ T cell function to immune control of Trypanosoma cruzi. Parasite Immunol. 1998, 20, 207–216.
- Sullivan, N.L.; Eickhoff, C.S.; Sagartz, J.; Hoft, D.F. Deficiency of antigenspecific B cells results in decreased Trypanosoma cruzi systemic but not mucosal immunity due to CD8 T cell exhaustion. J. Immunol. 2015, 194, 1806–1818.
- Andrade, D.V.; Gollob, K.J.; Dutra, W.O. Acute Chagas disease: New global challenges for an old neglected disease. PLoS Negl. Trop. Dis. 2014, 8, e3010.
- Ramstead, A.G.; Robison, A.; Blackwell, A.; Jerome, M.; Freedman, B.; Lubick, K.J.; Hedges, J.F.; Jutila, M.A. Roles of Toll-Like Receptor 2 (TLR2), TLR4, and MyD88 During Pulmonary Coxiella burnetii Infection. Infect. Immun. 2016, 84, 940–949.
- Torina, A.; Blanda, V.; Villari, S.; Piazza, A.; La Russa, F.; Grippi, F.; La Manna, M.P.; Di Liberto, D.; de la Fuente, J.; Sireci, G. Immune Response to Tick-Borne Hemoparasites: Host Adaptive Immune Response Mechanisms as Potential Targets for Therapies and Vaccines. Int. J. Mol. Sci. 2020, 21, 8813.
- Sireci, G.; Badami, G.D.; Di Liberto, D.; Blanda, V.; Grippi, F.; Di Paola, L.; Guercio, A.; de la Fuente, J.; Torina, A. Recent Advances on the Innate Immune Response to Coxiella burnetii. Front. Cell. Infect. Microbiol. 2021, 11, 754455.
- Pellegrini, A.; Guiñazu, N.; Giordanengo, L.; Cano, R.C.; Gea, S. The role of Toll-like receptors and adaptive immunity in the development of protective or pathological immune response triggered by the Trypanosoma cruzi protozoan. Future Microbiol. 2011, 6, 1521–1533.
- Campos, M.A.; Gazzinelli, R.T. Trypanosoma cruzi and its components as exogenous mediators of inflammation recognized through Toll-like receptors. Mediat. Inflamm. 2004, 13, 139–143.
- Tarleton, R.L. Immune system recognition of Trypanosoma cruzi. Curr. Opin. Immunol. 2007, 19, 430–434.
- Kayama, H.; Takeda, K. The innate immune response to Trypanosoma cruzi infection. Microbes Infect. 2010, 12, 511–517.
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384.
- Carrera-Silva, E.A.; Guinazu, N.; Pellegrini, A.; Cano, R.C.; Arocena, A.; Aoki, M.P.; Gea, S. Importance of TLR2 on hepatic immune and non-immune cells to attenuate the strong inflammatory liver response during Trypanosoma cruzi acute infection. PLoS Negl. Trop. Dis. 2010, 4, e863.
- Cerbán, F.M.; Stempin, C.C.; Volpini, X.; Carrera Silva, E.A.; Gea, S.; Motran, C.C. Signaling pathways that regulate Trypanosoma cruzi infection and immune response. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165707.
- Maganto-Garcia, E.; Punzon, C.; Terhorst, C.; Fresno, M. Rab5 activation by Toll-like receptor 2 is required for Trypanosoma cruzi internalization and replication in macrophages. Traffic 2008, 9, 1299–1315.
- Carrera-Silva, E.A.; Carolina, C.R.; Natalia, G.; Pilar, A.M.; Andrea, P.; Gea, S. TLR2, TLR4 and TLR9 are differentially modulated in liver lethally injured from BALB/c and C57BL/6 mice during Trypanosoma cruzi acute infection. Mol. Immunol. 2008, 45, 3580–3588.
- Campos, M.A.; Almeida, I.C.; Takeuchi, O.; Akira, S.; Valente, E.P.; Procópio, D.O.; Travassos, L.R.; Smith, J.A.; Golenbock, D.T.; Gazzinelli, R.T. Activation of toll-like receptor-2 by glycosylphosphatidylinositol anchors from a protozoan parasite. J. Immunol. 2001, 167, 416–423.
- Tarleton, R.L. CD8+ T cells in Trypanosoma cruzi infection. Semin. Immunopathol. 2015, 37, 233–238.
- da Costa, T.A.; Silva, M.V.; Mendes, M.T.; Carvalho-Costa, T.M.; Batista, L.R.; Lages-Silva, E.; Rodrigues, V.; Oliveira, C.J.; Ramirez, L.E. Immunomodulation by Trypanosoma cruzi: Toward understanding the association of dendritic cells with infecting TcI and TcII populations. J. Immunol. Res. 2014, 2014, 962047.
- Van Overtvelt, L.; Vanderheyde, N.; Verhasselt, V.; Ismaili, J.; De Vos, L.; Goldman, M.; Willems, F.; Vray, B. Trypanosoma cruzi infects human dendritic cells and prevents their maturation: Inhibition of cytokines, HLA-DR, and costimulatory molecules. Infect. Immun. 1999, 67, 4033–4040.
- Sathler-Avelar, R.; Lemos, E.M.; Reis, D.D.; Medrano-Mercado, N.; Araújo-Jorge, T.C.; Antas, P.R.; Corrêa-Oliveira, R.; Teixeira-Carvalho, A.; Elói-Santos, S.M.; Favato, D.; et al. Phenotypic features of peripheral blood leucocytes during early stages of human infection with Trypanosoma cruzi. Scand. J. Immunol. 2003, 58, 655–663.
- Dutra, W.O.; Menezes, C.A.; Villani, F.N.; da Costa, G.C.; da Silveira, A.B.; Reis, D.d.; Gollob, K.J. Cellular and genetic mechanisms involved in the generation of protective and pathogenic immune responses in human Chagas disease. Memórias Inst. Oswaldo Cruz 2009, 104, 208–218.
- Souza, P.E.; Rocha, M.O.; Menezes, C.A.; Coelho, J.S.; Chaves, A.C.; Gollob, K.J.; Dutra, W.O. Trypanosoma cruzi infection induces differential modulation of costimulatory molecules and cytokines by monocytes and T cells from patients with indeterminate and cardiac Chagas’ disease. Infect. Immun. 2007, 75, 1886–1894.
- de Araújo, F.F.; Corrêa-Oliveira, R.; Rocha, M.O.; Chaves, A.T.; Fiuza, J.A.; Fares, R.C.; Ferreira, K.S.; Nunes, M.C.; Keesen, T.S.; Damasio, M.P.; et al. Foxp3+CD25(high) CD4+ regulatory T cells from indeterminate patients with Chagas disease can suppress the effector cells and cytokines and reveal altered correlations with disease severity. Immunobiology 2012, 217, 768–777.
- Ropert, C.; Ferreira, L.R.; Campos, M.A.; Procópio, D.O.; Travassos, L.R.; Ferguson, M.A.; Reis, L.F.; Teixeira, M.M.; Almeida, I.C.; Gazzinelli, R.T. Macrophage signaling by glycosylphosphatidylinositol-anchored mucin-like glycoproteins derived from Trypanosoma cruzi trypomastigotes. Microbes Infect. 2002, 4, 1015–1025.
- Oliveira, A.C.; Peixoto, J.R.; de Arruda, L.B.; Campos, M.A.; Gazzinelli, R.T.; Golenbock, D.T.; Akira, S.; Previato, J.O.; Mendonça-Previato, L.; Nobrega, A.; et al. Expression of functional TLR4 confers proinflammatory responsiveness to Trypanosoma cruzi glycoinositolphospholipids and higher resistance to infection with T. cruzi. J. Immunol. 2004, 173, 5688–5696.
- Coelho, P.S.; Klein, A.; Talvani, A.; Coutinho, S.F.; Takeuchi, O.; Akira, S.; Silva, J.S.; Canizzaro, H.; Gazzinelli, R.T.; Teixeira, M.M. Glycosylphosphatidylinositol-anchored mucin-like glycoproteins isolated from Trypanosoma cruzi trypomastigotes induce in vivo leukocyte recruitment dependent on MCP-1 production by IFN-gamma-primed-macrophages. J. Leukoc. Biol. 2002, 71, 837–844.
- Stahl, P.; Schwarz, R.T.; Debierre-Grockiego, F.; Meyer, T. Trypanosoma cruzi parasites fight for control of the JAK-STAT pathway by disarming their host. JAKSTAT 2015, 3, e1012964.
- Sousa-Rocha, D.; Thomaz-Tobias, M.; Diniz, L.F.A.; Souza, P.S.S.; Pinge-Filho, P.; Toledo, K.A. Trypanosoma cruzi and Its Soluble Antigens Induce NET Release by Stimulating Toll-Like Receptors. PLoS ONE 2015, 10, e0139569.
- Cronemberger-Andrade, A.; Xander, P.; Soares, R.P.; Pessoa, N.L.; Campos, M.A.; Ellis, C.C.; Grajeda, B.; Ofir-Birin, Y.; Almeida, I.C.; Regev-Rudzki, N.; et al. Trypanosoma cruzi-Infected Human Macrophages Shed Proinflammatory Extracellular Vesicles That Enhance Host-Cell Invasion via Toll-Like Receptor 2. Front. Cell. Infect. Microbiol. 2020, 10, 99.
- Castillo, C.; Muñoz, L.; Carrillo, I.; Liempi, A.; Medina, L.; Galanti, N.; Maya, J.D.; Kemmerling, U. Toll-like receptor-2 mediates local innate immune response against Trypanosoma cruzi in ex vivo infected human placental chorionic villi explants. Placenta 2017, 60, 40–46.
- Blanda, V.; Bracale, U.M.; Di Taranto, M.D.; Fortunato, G. Galectin-3 in Cardiovascular Diseases. Int. J. Mol. Sci. 2020, 21, 9232.
- Brown, S.P.; Cornforth, D.M.; Mideo, N. Evolution of virulence in opportunistic pathogens: Generalism, plasticity, and control. Trends. Microbiol. 2012, 20, 336–342.
- Epting, C.L.; Coates, B.M.; Engman, D.M. Molecular mechanisms of host cell invasion by Trypanosoma cruzi. Exp. Parasitol. 2010, 126, 283–291.
- Koo, S.-J.; Szczesny, B.; Wan, X.; Putluri, N.; Garg, N.J. Pentose Phosphate Shunt Modulates Reactive Oxygen Species and Nitric Oxide Production Controlling Trypanosoma cruzi in Macrophages. Front. Immunol. 2018, 9, 202.
- Mesıas, A.C.; Garg, N.J.; Zago, M.P. Redox Balance Keepers and Possible Cell Functions Managed by Redox Homeostasis in Trypanosoma cruzi. Front. Cell. Infect. Microbiol. 2019, 9, 435.
- Piacenza, L.; Peluffo, G.; Alvarez, M.N.; Martınez, A.; Radi, R. Trypanosoma cruzi antioxidant enzymes as virulence factors in chagas disease. Antioxid. Redox Signal. 2013, 19, 723–734.
- Kipnis, T.L.; David, J.R.; Alper, C.A.; Sher, A.; da Silva, W.D. Enzymatic treatment transforms trypomastigotes of Trypanosoma cruzi into activators of alternative complement pathway and potentiates their uptake by macrophages. Proc. Natl. Acad. Sci. USA 1981, 78, 602–605.
- Norris, K.A.; Bradt, B.; Cooper, N.R.; So, M. Characterization of a Trypanosoma cruzi C3 binding protein with functional and genetic similarities to the human complement regulatory protein, decay-accelerating factor. J. Immunol. 1991, 147, 2240–2247.
- Tambourgi, D.V.; Kipnis, T.L.; da Silva, W.D.; Joiner, K.A.; Sher, A.; Heath, S.; Hall, B.F.; Ogden, G.B. A partial cDNA clone of trypomastigote decay-accelerating factor (T-DAF), a developmentally regulated complement inhibitor of Trypanosoma cruzi, has genetic and functional similarities to the human complement inhibitor DAF. Infect. Immun. 1993, 61, 3656–3663.
- Schenkman, S.; Eichinger, D.; Pereira, M.E.A.; Nussenzweig, V. Structural and functional properties of Trypanosoma trans-sialidase. Annu. Rev. Microbiol. 1994, 48, 499–523.
- Valck, C.; Ramirez, G.; Lopez, N.; Ribeiro, C.H.; Maldonado, I.; Sanchez, G.; Ferreira, V.P.; Schwaeble, W.; Ferreira, A. Molecular mechanisms involved in the inactivation of the first component of human complement by Trypanosoma cruzi calreticulin. Mol. Immunol. 2010, 47, 1516–1521.
- Chamond, N.; Gregoire, C.; Coatnoan, N.; Rougeot, C.; Freitas Junior, L.H.; da Silveira, J.F.; Degrave, W.M.; Minoprio, P. Biochemical characterization of proline racemases from the human protozoan parasite Trypanosoma cruzi and definition of putative protein signatures. J. Biol. Chem. 2003, 278, 15484–15494.
- Reina-San-Martin, B.; Degrave, W.; Rougeot, C.; Cosson, A.; Chamond, N.; Cordeiro-Da-Silva, A.; Arala-Chaves, M.; Coutinho, A.; Minoprio, P. A B-cell mitogen from a pathogenic trypanosome is a eukaryotic proline racemase. Nat. Med. 2000, 6, 890–897.
- Chamond, N.; Goytia, M.; Coatnoan, N.; Barale, J.C.; Cosson, A.; Degrave, W.M.; Minoprio, P. Trypanosoma cruzi proline racemases are involved in parasite differentiation and infectivity. Mol. Microbiol. 2005, 58, 46–60.
- Ouaissi, M.A.; Dubremetz, J.F.; Schoneck, R.; Fernandez-Gomez, R.; Gomez-Corvera, R.; Billaut-Mulot, O.; Taibi, A.; Loyens, M.; Tartar, A.; Sergheraert, C.; et al. Trypanosoma cruzi: A 52-kDa protein sharing sequence homology with glutathione S-transferase is localized in parasite organelles morphologically resembling reservosomes. Exp. Parasitol. 1995, 81, 453–461.
- Ouaissi, M.A.; Guilvard, E.; Delneste, Y.; Caron, G.; Magistrelli, G.; Herbault, N.; Thieblemont, N.; Jeannin, P. The Trypanosoma cruzi Tc52- released protein induces human dendritic cell maturation, signals via toll-like receptor 2, and confers protection against lethal infection. J. Immunol. 2002, 168, 6366–6374.
- Bonfim-Melo, A.; Ferreira, E.R.; Florentino, P.T.V.; Mortara, R.A. Amastigote Synapse: The Tricks of Trypanosoma cruzi Extracellular Amastigotes. Front. Microbiol. 2018, 9, 1341.
- Rodrigues, A.A.; Clemente, T.M.; Dos Santos, M.A.; Machado, F.C.; Gomes, R.G.; Moreira, H.H.; Cruz, M.C.; Brígido, P.C.; Dos Santos, P.C.; Martins, F.A.; et al. A Recombinant Protein Based on Trypanosoma cruzi P21 Enhances Phagocytosis. PLoS ONE 2012, 7, e51384.
- Ferreira, É.R.; Horjales, E.; Bonfim-Melo, A.; Cortez, C.; da Silva, C.V.; De Groote, M.; Sobreira, T.J.P.; Cruz, M.C.; Lima, F.M.; Cordero, E.M.; et al. Unique behavior of Trypanosoma cruzi mevalonate kinase: A conserved glycosomal enzyme involved in host cell invasion and signaling. Sci. Rep. 2016, 6, 24610.
- de Castro Neto, A.L.; da Silveira, J.F.; Mortara, R.A. Comparative Analysis of Virulence Mechanisms of Trypanosomatids Pathogenic to Humans. Front. Cell. Infect. Microbiol. 2021, 16, 669079.
- Freire-De-Lima, L.; Fonseca, L.M.; Oeltmann, T.; Mendoncą-Previato, L.; Previato, J.O. The trans-sialidase, the major Trypanosoma cruzi virulence factor: Three decades of studies. Glycobiology 2015, 25, 1142–1149.
More