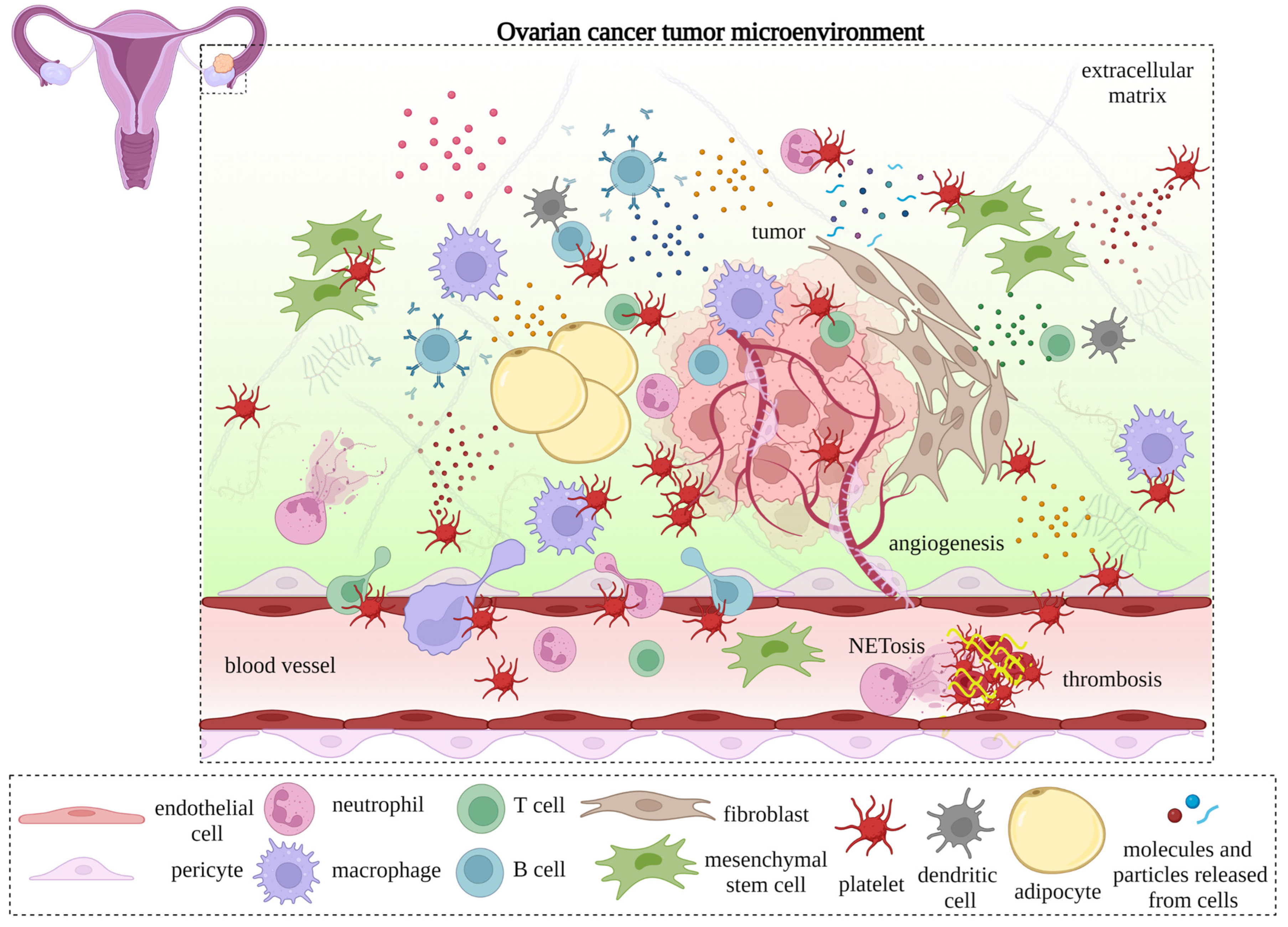
Platelets, the primary operatives of hemostasis that contribute to blood coagulation and wound healing after blood vessel injury, are also involved in pathological conditions, including cancer. Malignancy-associated thrombosis is common in ovarian cancer patients and is associated with poor clinical outcomes. Platelets extravasate into the tumor microenvironment (TME) in ovarian cancer and interact with cancer cells and non-cancerous elements. Ovarian cancer cells also activate platelets. The communication between activated platelets, cancer cells, and the tumor microenvironment is via various platelet membrane proteins or mediators released through degranulation or the secretion of microvesicles from platelets. These interactions trigger signaling cascades in tumors that promote ovarian cancer progression, metastasis, and neoangiogenesis.
- ovarian cancer
- platelet
- tumor microenvironment
- metastasis
- angiogenesis
- thrombosis
- immune system
1. Introduction
| Location | General Function | Examples | References |
|---|---|---|---|
| Surface molecules |
Integrins | α2β1 (GPIa/IIa), α5β1, α6β1, αLβ2 (ICAM-2), αIIbβ3 (GPIIb/IIIa), αVβ3 | [36] |
| Selectins | P-selectin (CD62P), CLEC-2 | [37][38] | |
| Leucine-rich repeat receptors |
GPIb-IX-V, TLR1, TLR2, TLR4, TLR6, MMPs | [36][39][40] | |
| ADP receptors | P2Y1, P2Y12 | [41] | |
| Thrombin receptors | PAR1, PAR4, GPIbα | [42][43] | |
| Tetraspanins | CD63, CD9, CD53 | [36] | |
| Prostaglandin receptors | PGD2 and PGE2 receptors | [44] | |
| Prostacyclin receptors | PGI2 receptors | [44] | |
| Thromboxane receptors | TxA2 receptors | [45] | |
| Lipid receptors | PAF and LPA receptors | [46][47] | |
| Ig receptors | GPVI, FcγRIIA (CD32), FcεRI (CD23) | [48][49] | |
| JAMs | JAM-1, JAM-2, JAM-3, PECAM-1 (CD31) | [50][51] | |
| Tyrosine kinase receptors | Thrombopoietin, leptin, insulin, PDGF receptors | [36] | |
| Immune checkpoints | PD-L1, GITRL, OX40L | [52][53][54] | |
| Other receptors | Serotonin receptors, GPIV (CD36), IAP (CD47) complement receptors, CD40, CD40L (CD154) |
[55][56][57][58][59] | |
| α-granules | Adhesion molecules | vWF, αIIbβ3 (GPIIb/IIIa), αVβ3, P-selectin (CD62P), fibrinogen, fibronectin, thrombospondin | [36][60][61] |
| Proangiogenic factors | VEGF, angiopoietin-1, SDF-1 (CXCL12), S1P, TGF-β, IL-6, PF4 (CXCL4) | [61] | |
| Angiostatic factors | Endostatin, angiostatin, thrombospondin-1 | [56][61] | |
| Growth factors | VEGF, PDGF, EGF, FGF, HGF, IGF-1, CTGF, TGF-β | [61][62][63][64] | |
| Coagulation-associated components | Prothrombin, fibrinogen, factor V, factor VIII, factor XI, protein S |
[61][65][66] | |
| Fibrinolytic factors | α2-macroglobulin, uPA, PAI-1 | [61] | |
| MMPs | MMP-1, MMP-2, MMP-3, MMP-9 | [67][68] | |
| Metalloproteinases | ADAM-10, ADAM-17, ADAMTS-13 | [69] | |
| TIMPs | TIMP-1, TIMP-2, TIMP-4 | [69] | |
| Inflammamodulatory molecules | CXCL1, PF4 (CXCL4), CXCL5, CXCL7 (NAP-2), IL-1β, IL-6, IL-8 (CXCL8), SDF-1 (CXCL12), CCL2 (MCP-1), CCL3 (MIP-1α), CCL5 (RANTES), CCL7, PAF, LPA, TGF-β, TNF-α, GM-CSF |
[61][70][71][72][73][74][75] | |
| Immunologic molecules | Complement factors, IgG, IgA, IgM, thymosin-β4 | [61][76][77][78] | |
| Other components | Albumin, α1-antitrypsin, HMWK |
[61] | |
| δ-granules | Nucleotides | ADP, ATP, GDP, GTP | [79] |
| Bioactive amines | Serotonin, histamine, epinephrine | [60] | |
| Ions | Calcium, magnesium, phosphate, pyrophosphate | [61] | |
| Polyphosphates | Polyphosphate (polyP) | [60] | |
| Lysosomes | Proteases | Cathepsin D/E, carboxypeptidase A/B, glycohydrolases, collagenase, elastase | [79][80] |
| Phosphatases | Acid phosphatase | [79] | |
| Phospholipases | Phospholipase A | [61] |
2. Interactions of Platelets with TME Compartments: Endothelial Cells, Pericytes, and Cancer-Associated Fibroblasts
2.1. Interactions with Endothelial Cells
2.1.1. In Angiogenesis

2.1.2. In Lymphangiogenesis
2.2. Interactions with Pericytes
2.3. Interactions with Cancer-Associated Fibroblasts
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics. CA Cancer J. Clin. 2022, 72, 7–33.
- Jelovac, D.; Armstrong, D.K. Recent progress in the diagnosis and treatment of ovarian cancer. CA Cancer J. Clin. 2011, 61, 183–203.
- Etemadmoghadam, D.; Defazio, A.; Beroukhim, R.; Mermel, C.; George, J.; Getz, G.; Tothill, R.; Okamoto, A.; Raeder, M.B.; Harnett, P.; et al. Integrated Genome-Wide DNA Copy Number and Expression Analysis Identifies Distinct Mechanisms of Primary Chemoresistance in Ovarian Carcinomas. Clin. Cancer Res. 2009, 15, 1417–1427.
- Patch, A.-M.; Christie, E.L.; Etemadmoghadam, D.; Garsed, D.W.; George, J.; Fereday, S.; Nones, K.; Cowin, P.; Alsop, K.; Bailey, P.J.; et al. Whole–genome characterization of chemoresistant ovarian cancer. Nature 2015, 521, 489–494.
- Jiménez-Sánchez, A.; Cybulska, P.; Mager, K.L.; Koplev, S.; Cast, O.; Couturier, D.-L.; Memon, D.; Selenica, P.; Nikolovski, I.; Mazaheri, Y.; et al. Unraveling tumor–immune heterogeneity in advanced ovarian cancer uncovers immunogenic effect of chemotherapy. Nat. Genet. 2020, 52, 582–593.
- Jin, M.-Z.; Jin, W.-L. The updated landscape of tumor microenvironment and drug repurposing. Signal Transduct. Target. Ther. 2020, 5, 166.
- Jordan, K.R.; Sikora, M.J.; Slansky, J.E.; Minic, A.; Richer, J.K.; Moroney, M.R.; Hu, J.; Wolsky, R.J.; Watson, Z.L.; Yamamoto, T.M.; et al. The Capacity of the Ovarian Cancer Tumor Microenvironment to Integrate Inflammation Signaling Conveys a Shorter Disease-free Interval. Clin. Cancer Res. 2020, 26, 6362–6373.
- CChen, F.; Zhuang, X.; Lin, L.; Yu, P.; Wang, Y.; Shi, Y.; Hu, G.; Sun, Y. New horizons in tumor microenvironment biology: Challenges and opportunities. BMC Med. 2015, 13, 2498.
- Giraldo, N.A.; Sanchez-Salas, R.; Peske, J.D.; Vano, Y.; Becht, E.; Petitprez, F.; Validire, P.; Ingels, A.; Cathelineau, X.; Fridman, W.H.; et al. The clinical role of the TME in solid cancer. Br. J. Cancer 2019, 120, 45–53.
- Corvigno, S.; Burks, J.K.; Hu, W.; Zhong, Y.; Jennings, N.B.; Fleming, N.D.; Westin, S.N.; Fellman, B.; Liu, J.; Sood, A.K. Immune microenvironment composition in high-grade serous ovarian cancers based on BRCA mutational status. J. Cancer Res. Clin. Oncol. 2021, 147, 3545–3555.
- Kreuzinger, C.; Geroldinger, A.; Smeets, D.; Braicu, E.I.; Sehouli, J.; Koller, J.; Wolf, A.; Darb-Esfahani, S.; Joehrens, K.; Vergote, I.; et al. A Complex Network of Tumor Microenvironment in Human High-Grade Serous Ovarian Cancer. Clin. Cancer Res. 2017, 23, 7621–7632.
- Ukita, M.; Hamanishi, J.; Yoshitomi, H.; Yamanoi, K.; Takamatsu, S.; Ueda, A.; Suzuki, H.; Hosoe, Y.; Furutake, Y.; Taki, M.; et al. CXCL13-producing CD4+ T cells accumulate in the early phase of tertiary lymphoid structures in ovarian cancer. J. Clin. Investig. 2022, 7, e157215.
- Taylor, S.E.; Chan, D.K.; Yang, D.; Bruno, T.; Lieberman, R.; Siddiqui, J.; Soong, T.R.; Coffman, L.; Buckanovich, R.J. Shifting the Soil: Metformin Treatment Decreases the Protumorigenic Tumor Microenvironment in Epithelial Ovarian Cancer. Cancers 2022, 14, 2298.
- Cho, M.S.; Lee, H.; Gonzalez-Delgado, R.; Li, D.; Sasano, T.; Carlos-Alcalde, W.; Ma, Q.; Liu, J.; Sood, A.K.; Afshar-Kharghan, V. Platelets Increase the Expression of PD-L1 in Ovarian Cancer. Cancers 2022, 14, 2498.
- Miyashita, T.; Tajima, H.; Gabata, R.; Okazaki, M.; Shimbashi, H.; Ohbatake, Y.; Okamoto, K.; Nakanuma, S.; Sakai, S.; Makino, I.; et al. Impact of Extravasated Platelet Activation and Podoplanin-positive Cancer-associated Fibroblasts in Pancreatic Cancer Stroma. Anticancer. Res. 2019, 39, 5565–5572.
- Nasti, T.H.; Bullard, D.C.; Yusuf, N. P-selectin enhances growth and metastasis of mouse mammary tumors by promoting regulatory T cell infiltration into the tumors. Life Sci. 2015, 131, 11–18.
- Yamaguchi, T.; Fushida, S.; Kinoshita, J.; Okazaki, M.; Ishikawa, S.; Ohbatake, Y.; Terai, S.; Okamoto, K.; Nakanuma, S.; Makino, I.; et al. Extravasated platelet aggregation contributes to tumor progression via the accumulation of myeloid-derived suppressor cells in gastric cancer with peritoneal metastasis. Oncol. Lett. 2020, 20, 1879–1887.
- Lonsdorf, A.S.; Krämer, B.F.; Fahrleitner, M.; Schönberger, T.; Gnerlich, S.; Ring, S.; Gehring, S.; Schneider, S.W.; Kruhlak, M.J.; Meuth, S.G.; et al. Engagement of αIIbβ3 (GPIIb/IIIa) with ανβ3 Integrin Mediates Interaction of Melanoma Cells with Platelets. J. Biol. Chem. 2012, 287, 2168–2178.
- Shirai, T.; Inoue, O.; Tamura, S.; Tsukiji, N.; Sasaki, T.; Endo, H.; Satoh, K.; Osada, M.; Sato-Uchida, H.; Fujii, H.; et al. C-type lectin-like receptor 2 promotes hematogenous tumor metastasis and prothrombotic state in tumor-bearing mice. J. Thromb. Haemost. 2016, 15, 513–525.
- Haemmerle, M.; Bottsford-Miller, J.; Pradeep, S.; Taylor, M.L.; Choi, H.-J.; Hansen, J.M.; Dalton, H.J.; Stone, R.L.; Cho, M.S.; Nick, A.M.; et al. FAK regulates platelet extravasation and tumor growth after antiangiogenic therapy withdrawal. J. Clin. Investig. 2016, 126, 1885–1896.
- Joseph, R.; Soundararajan, R.; Vasaikar, S.; Yang, F.; Allton, K.L.; Tian, L.; Hollander, P.D.; Isgandarova, S.; Haemmerle, M.; Mino, B.; et al. CD8+ T cells inhibit metastasis and CXCL4 regulates its function. Br. J. Cancer 2021, 125, 176–189.
- Plantureux, L.; Mège, D.; Crescence, L.; Carminita, E.; Robert, S.; Cointe, S.; Brouilly, N.; Ezzedine, W.; Dignat-George, F.; Dubois, C.; et al. The Interaction of Platelets with Colorectal Cancer Cells Inhibits Tumor Growth but Promotes Metastasis. Cancer Res. 2020, 80, 291–303.
- Pavlovic, N.; Rani, B.; Gerwins, P.; Heindryckx, F. Platelets as Key Factors in Hepatocellular Carcinoma. Cancers 2019, 11, 1022.
- Levoux, J.; Prola, A.; Lafuste, P.; Gervais, M.; Chevallier, N.; Koumaiha, Z.; Kefi, K.; Braud, L.; Schmitt, A.; Yacia, A.; et al. Platelets Facilitate the Wound-Healing Capability of Mesenchymal Stem Cells by Mitochondrial Transfer and Metabolic Reprogramming. Cell Metab. 2021, 33, 283–299.
- Sibilano, M.; Tullio, V.; Adorno, G.; Savini, I.; Gasperi, V.; Catani, M.V. Platelet-Derived miR-126-3p Directly Targets AKT2 and Exerts Anti-Tumor Effects in Breast Cancer Cells: Further Insights in Platelet-Cancer Interplay. Int. J. Mol. Sci. 2022, 23, 5484.
- Ye, B.; Li, F.; Chen, M.; Weng, Y.; Qi, C.; Xie, Y.; Zhang, Q.; Ding, H.; Zhang, J.; Gao, X. A panel of platelet-associated circulating long non-coding RNAs as potential biomarkers for colorectal cancer. Genomics 2021, 114, 31–37.
- Stone, R.L.; Nick, A.M.; McNeish, I.A.; Balkwill, F.; Han, H.D.; Bottsford-Miller, J.; Rupaimoole, R.; Armaiz-Pena, G.N.; Pecot, C.V.; Coward, J.; et al. Paraneoplastic Thrombocytosis in Ovarian Cancer. N. Engl. J. Med. 2012, 366, 610–618.
- Allensworth, S.; Langstraat, C.; Martin, J.; Lemens, M.; McGree, M.; Weaver, A.; Dowdy, S.; Podratz, K.; Bakkum-Gamez, J. Evaluating the prognostic significance of preoperative thrombocytosis in epithelial ovarian cancer. Gynecol. Oncol. 2013, 130, 499–504.
- Cohen, J.G.; Tran, A.-Q.; Rimel, B.; Cass, I.; Walsh, C.S.; Karlan, B.Y.; Li, A.J. Thrombocytosis at secondary cytoreduction for recurrent ovarian cancer predicts suboptimal resection and poor survival. Gynecol. Oncol. 2014, 132, 556–559.
- Bottsford-Miller, J.; Choi, H.-J.; Dalton, H.J.; Stone, R.L.; Cho, M.S.; Haemmerle, M.; Nick, A.M.; Pradeep, S.; Zand, B.; Previs, R.A.; et al. Differential Platelet Levels Affect Response to Taxane-Based Therapy in Ovarian Cancer. Clin. Cancer Res. 2015, 21, 602–610.
- Cho, M.S.; Bottsford-Miller, J.; Vasquez, H.G.; Stone, R.; Zand, B.; Kroll, M.H.; Sood, A.K.; Afshar-Kharghan, V. Platelets increase the proliferation of ovarian cancer cells. Blood 2012, 120, 4869–4872.
- Guo, Y.; Cui, W.; Pei, Y.; Xu, D. Platelets promote invasion and induce epithelial to mesenchymal transition in ovarian cancer cells by TGF-β signaling pathway. Gynecol. Oncol. 2019, 153, 639–650.
- Haemmerle, M.; Taylor, M.L.; Gutschner, T.; Pradeep, S.; Cho, M.S.; Sheng, J.; Lyons, Y.M.; Nagaraja, A.S.; Dood, R.L.; Wen, Y.; et al. Platelets reduce anoikis and promote metastasis by activating YAP1 signaling. Nat. Commun. 2017, 8, 310.
- Davis, A.N.; Afshar-Kharghan, V.; Sood, A.K. Platelet Effects on Ovarian Cancer. Semin. Oncol. 2014, 41, 378–384.
- Cho, M.S.; Gonzalez-Pagan, O.; Pinto, K.C.; Sood, A.; Afshar-Kharghan, V. The Inhibition of Platelets Restore Anti-Tumor Immune Response to Ovarian Cancer and Its Therapeutic Implication. Blood 2018, 132, 3698.
- Saboor, M.; Ayub, Q.; Ilyas, S. Moinuddin Platelet receptors; an instrumental of platelet physiology. Pak. J. Med. Sci. 2013, 29, 891–896.
- McEver, R.P.; Beckstead, J.H.; Moore, K.L.; Marshall-Carlson, L.; Bainton, D.F. GMP-140, a platelet alpha-granule membrane protein, is also synthesized by vascular endothelial cells and is localized in Weibel-Palade bodies. J. Clin. Investig. 1989, 84, 92–99.
- Rayes, J.; Lax, S.; Wichaiyo, S.; Watson, S.K.; Di, Y.; Lombard, S.; Grygielska, B.; Smith, S.W.; Skordilis, K.; Watson, S.P. The podoplanin-CLEC-2 axis inhibits inflammation in sepsis. Nat. Commun. 2017, 8, 2239.
- Cognasse, F.; Nguyen, K.A.; Damien, P.; McNicol, A.; Pozzetto, B.; Hamzeh-Cognasse, H.; Garraud, O. The inflammatory role of platelets via their TLRs and Siglec receptors. Front. Immunol. 2015, 6, 83.
- Clark, S.R.; Ma, A.C.; Tavener, S.A.; McDonald, B.; Goodarzi, Z.; Kelly, M.M.; Patel, K.D.; Chakrabarti, S.; McAvoy, E.; Sinclair, G.D.; et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat. Med. 2007, 13, 463–469.
- Cho, M.S.; Noh, K.; Haemmerle, M.; Li, D.; Park, H.; Hu, Q.; Hisamatsu, T.; Mitamura, T.; Mak, S.L.C.; Kunapuli, S.; et al. Role of ADP receptors on platelets in the growth of ovarian cancer. Blood 2017, 130, 1235–1242.
- Camerer, E.; Qazi, A.A.; Duong, D.N.; Cornelissen, I.; Advincula, R.; Coughlin, S.R. Platelets, protease-activated receptors, and fibrinogen in hematogenous metastasis. Blood 2004, 104, 397–401.
- Jain, S.; Zuka, M.; Liu, J.; Russell, S.; Dent, J.; Guerrero, J.A.; Forsyth, J.; Maruszak, B.; Gartner, T.K.; Felding-Habermann, B.; et al. Platelet glycoprotein Ibα supports experimental lung metastasis. Proc. Natl. Acad. Sci. USA 2007, 104, 9024–9028.
- Tynan, S.S.; Andersen, N.H.; Wills, M.T.; Harker, L.A.; Hanson, S.R. On the multiplicity of platelet prostaglandin receptors. Prostaglandins 1984, 27, 683–696.
- Paul, B.Z.S.; Jin, J.; Kunapuli, S.P. Molecular Mechanism of Thromboxane A2-induced Platelet Aggregation. J. Biol. Chem. 1999, 274, 29108–29114.
- Burgers, J.A.; Akkerman, J.W.N. Regulation of the receptor for platelet-activating factor on human platelets. Biochem. J. 1993, 291, 157–161.
- Motohashi, K.; Shibata, S.; Ozaki, Y.; Yatomi, Y.; Igarashi, Y. Identification of lysophospholipid receptors in human platelets: The relation of two agonists, lysophosphatidic acid and sphingosine 1-phosphate. FEBS Lett. 2000, 468, 189–193.
- Tsuji, M.; Ezumi, Y.; Arai, M.; Takayama, H. A Novel Association of Fc Receptor γ-Chain with Glycoprotein VI and Their Co-expression as a Collagen Receptor in Human Platelets. J. Biol. Chem. 1997, 272, 23528–23531.
- Hasegawa, S.; Pawankar, R.; Suzuki, K.; Nakahata, T.; Furukawa, S.; Okumura, K.; Ra, C. Functional expression of the high affinity receptor for IgE (FcepsilonRI) in human platelets and its’ intracellular expression in human megakaryocytes. Blood 1999, 93, 2543–2551.
- Santoso, S.; Sachs, U.J.H.; Kroll, H.; Linder, M.; Ruf, A.; Preissner, K.T.; Chavakis, T. The Junctional Adhesion Molecule 3 (JAM-3) on Human Platelets is a Counterreceptor for the Leukocyte Integrin Mac-1. J. Exp. Med. 2002, 196, 679–691.
- Newman, D.K.; Hamilton, C.; Newman, P.J. Inhibition of antigen-receptor signaling by Platelet Endothelial Cell Adhesion Molecule-1 (CD31) requires functional ITIMs, SHP-2, and p56lck. Blood 2001, 97, 2351–2357.
- Hinterleitner, C.; Strähle, J.; Malenke, E.; Hinterleitner, M.; Henning, M.; Seehawer, M.; Bilich, T.; Heitmann, J.; Lutz, M.; Mattern, S.; et al. Platelet PD-L1 reflects collective intratumoral PD-L1 expression and predicts immunotherapy response in non-small cell lung cancer. Nat. Commun. 2021, 12, 7005.
- Zhou, Y.; Heitmann, J.S.; Clar, K.L.; Kropp, K.N.; Hinterleitner, M.; Engler, T.; Koch, A.; Hartkopf, A.D.; Zender, L.; Salih, H.R.; et al. Platelet-expressed immune checkpoint regulator GITRL in breast cancer. Cancer Immunol. Immunother. 2021, 70, 2483–2496.
- Rittig, S.M.; Lutz, M.S.; Clar, K.L.; Zhou, Y.; Kropp, K.N.; Koch, A.; Hartkopf, A.D.; Hinterleitner, M.; Zender, L.; Salih, H.R.; et al. Controversial Role of the Immune Checkpoint OX40L Expression on Platelets in Breast Cancer Progression. Front. Oncol. 2022, 12, 917834.
- Dale, G.L.; Friese, P.; Batar, P.; Hamilton, S.F.; Reed, G.L.; Jackson, K.W.; Clemetson, K.J.; Alberio, L. Stimulated platelets use serotonin to enhance their retention of procoagulant proteins on the cell surface. Nature 2002, 415, 175–179.
- Isenberg, J.S.; Romeo, M.J.; Yu, C.; Nghiem, K.; Monsale, J.; Rick, M.E.; Wink, D.A.; Frazier, W.A.; Roberts, D.D. Thrombospondin-1 stimulates platelet aggregation by blocking the antithrombotic activity of nitric oxide/cGMP signaling. Blood 2008, 111, 613–623.
- Hamzeh-Cognasse, H.; Damien, P.; Chabert, A.; Pozzetto, B.; Cognasse, F.; Garraud, O. Platelets and Infections-Complex Interactions with Bacteria. Front. Immunol. 2015, 6, 82.
- Inwald, D.P.; McDowall, A.; Peters, M.J.; Callard, R.E.; Klein, N.J. CD40 Is Constitutively Expressed on Platelets and Provides a Novel Mechanism for Platelet Activation. Circ. Res. 2003, 92, 1041–1048.
- Huang, J.; Jochems, C.; Talaie, T.; Anderson, A.; Jales, A.; Tsang, K.Y.; Madan, R.A.; Gulley, J.L.; Schlom, J. Elevated serum soluble CD40 ligand in cancer patients may play an immunosuppressive role. Blood 2012, 120, 3030–3038.
- Fitch-Tewfik, J.L.; Flaumenhaft, R. Platelet Granule Exocytosis: A Comparison with Chromaffin Cells. Front. Endocrinol. 2013, 4, 77.
- Wojtukiewicz, M.Z.; Sierko, E.; Hempel, D.; Tucker, S.C.; Honn, K.V. Platelets and cancer angiogenesis nexus. Cancer Metastasis Rev. 2017, 36, 249–262.
- Kim, S.; Garcia, A.; Jackson, S.P.; Kunapuli, S.P. Insulin-like growth factor-1 regulates platelet activation through PI3-Kα isoform. Blood 2007, 110, 4206–4213.
- Kubota, S.; Kawata, K.; Yanagita, T.; Doi, H.; Kitoh, T.; Takigawa, M. Abundant Retention and Release of Connective Tissue Growth Factor (CTGF/CCN2) by Platelets. J. Biochem. 2004, 136, 279–282.
- Haemmerle, M.; Stone, R.L.; Menter, D.G.; Afshar-Kharghan, V.; Sood, A.K. The Platelet Lifeline to Cancer: Challenges and Opportunities. Cancer Cell 2018, 33, 965–983.
- Walsh, P.N. Platelets and coagulation proteins. Fed. Proc. 1981, 40, 2086–2091.
- Stavenuiter, F.; Davis, N.F.; Duan, E.; Gale, A.J.; Heeb, M.J. Platelet protein S directly inhibits procoagulant activity on platelets and microparticles. Thromb. Haemost. 2013, 109, 229–237.
- Trivedi, V.; Boire, A.; Tchernychev, B.; Kaneider, N.C.; Leger, A.J.; O’Callaghan, K.; Covic, L.; Kuliopulos, A. Platelet Matrix Metalloprotease-1 Mediates Thrombogenesis by Activating PAR1 at a Cryptic Ligand Site. Cell 2009, 137, 332–343.
- Sheu, J.R.; Fong, T.H.; Liu, C.M.; Shen, M.Y.; Chen, T.L.; Chang, Y.; Lu, M.S.; Hsiao, G. Expression of matrix metalloproteinase-9 in human platelets: Regulation of platelet activation in in vitro and in vivo studies. Br. J. Pharmacol. 2004, 143, 193–201.
- Santos-Martínez, M.J.; Medina, C.; Jurasz, P.; Radomski, M.W. Role of metalloproteinases in platelet function. Thromb. Res. 2008, 121, 535–542.
- Lishko, V.K.; Yakubenko, V.P.; Ugarova, T.P.; Podolnikova, N.P. Leukocyte integrin Mac-1 (CD11b/CD18, αMβ2, CR3) acts as a functional receptor for platelet factor 4. J. Biol. Chem. 2018, 293, 6869–6882.
- Walz, A.; Dewald, B.; von Tscharner, V.; Baggiolini, M. Effects of the neutrophil-activating peptide NAP-2, platelet basic protein, connective tissue-activating peptide III and platelet factor 4 on human neutrophils. J. Exp. Med. 1989, 170, 1745–1750.
- Chen, R.; Jin, G.; Li, W.; McIntyre, T.M. Epidermal Growth Factor (EGF) Autocrine Activation of Human Platelets Promotes EGF Receptor–Dependent Oral Squamous Cell Carcinoma Invasion, Migration, and Epithelial Mesenchymal Transition. J. Immunol. 2018, 201, 2154–2164.
- Chen, Y.; Zhong, H.; Zhao, Y.; Luo, X.; Gao, W. Role of platelet biomarkers in inflammatory response. Biomark. Res. 2020, 8, 28.
- Bock, M.; Bergmann, C.B.; Jung, S.; Kalbitz, M.; Relja, B.; Huber-Wagner, S.; Biberthaler, P.; van Griensven, M.; Hanschen, M. The posttraumatic activation of CD4+ T regulatory cells is modulated by TNFR2- and TLR4-dependent pathways, but not by IL-10. Cell. Immunol. 2018, 331, 137–145.
- Raiden, S.; Schettini, J.; Salamone, G.; Trevani, A.; Vermeulen, M.; Gamberale, R.; Giordano, M.; Geffner, J. Human Platelets Produce Granulocyte-Macrophage Colony-Stimulating Factor and Delay Eosinophil Apoptosis. Lab. Investig. 2003, 83, 589–598.
- Kim, H.; Conway, E.M. Platelets and Complement Crosstalk in Early Atherogenesis. Front. Cardiovasc. Med. 2019, 6, 131.
- George, J.N.; Saucerman, S. Platelet Igg, Iga, Igm, and Albumin: Correlation of Platelet and Plasma Concentrations in Normal Subjects and in Patients with Itp or Dysproteinemia. Blood 1988, 72, 362–365.
- Huff, T.; Otto, A.M.; Müller, C.S.G.; Meier, M.; Hannappel, E. Thymosin β4 is released from human blood platelets and attached by factor XIIIa (transglutaminase) to fibrin and collagen. FASEB J. 2002, 16, 691–696.
- de Jong, J.S.S.G.; Dekker, L.R.C. Platelets and Cardiac Arrhythmia. Front. Physiol. 2010, 1, 166.
- Chesney, C.M.; Harper, E.; Colman, R.W. Human Platelet Collagenase. J. Clin. Investig. 1974, 53, 1647–1654.
- Lugano, R.; Ramachandran, M.; Dimberg, A. Tumor angiogenesis: Causes, consequences, challenges and opportunities. Cell. Mol. Life Sci. 2020, 77, 1745–1770.
- Nagy, J.A.; Chang, S.-H.; Dvorak, A.M.; Dvorak, H.F. Why are tumour blood vessels abnormal and why is it important to know. Br. J. Cancer 2009, 100, 865–869.
- Klein, D. The Tumor Vascular Endothelium as Decision Maker in Cancer Therapy. Front. Oncol. 2018, 8, 367.
- Chaplin, D.J.; Olive, P.L.; Durand, R.E. Intermittent blood flow in a murine tumor: Radiobiological effects. Cancer Res. 1987, 47, 597–601.
- Fitzgerald, G.; Soro-Arnaiz, I.; de Bock, K. The Warburg Effect in Endothelial Cells and its Potential as an Anti-angiogenic Target in Cancer. Front. Cell Dev. Biol. 2018, 6, 100.
- Dvorak, H.F.; Brown, L.F.; Detmar, M.; Dvorak, A.M. Vascular permeability factor/vascular endothelial growth factor, microvascular hyperpermeability, and angiogenesis. Am. J. Pathol. 1995, 146, 1029–1039.
- Perren, T.J.; Swart, A.M.; Pfisterer, J.; Ledermann, J.A.; Pujade-Lauraine, E.; Kristensen, G.; Carey, M.S.; Beale, P.; Cervantes, A.; Kurzeder, C.; et al. A Phase 3 Trial of Bevacizumab in Ovarian Cancer. N. Engl. J. Med. 2011, 365, 2484–2496.
- Burger, R.A.; Sill, M.W.; Monk, B.J.; Greer, B.E.; Sorosky, J.I. Phase II Trial of Bevacizumab in Persistent or Recurrent Epithelial Ovarian Cancer or Primary Peritoneal Cancer: A Gynecologic Oncology Group Study. J. Clin. Oncol. 2007, 25, 5165–5171.
- Choi, H.-J.; Pena, G.N.A.; Pradeep, S.; Cho, M.S.; Coleman, R.L.; Sood, A.K. Anti-vascular therapies in ovarian cancer: Moving beyond anti-VEGF approaches. Cancer Metastasis Rev. 2014, 34, 19–40.
- Kisucka, J.; Butterfield, C.E.; Duda, D.G.; Eichenberger, S.C.; Saffaripour, S.; Ware, J.; Ruggeri, Z.M.; Jain, R.K.; Folkman, J.; Wagner, D.D. Platelets and platelet adhesion support angiogenesis while preventing excessive hemorrhage. Proc. Natl. Acad. Sci. USA 2006, 103, 855–860.
- Martins, P.D.C.; García-Vallejo, J.-J.; van Thienen, J.V.; Fernandez-Borja, M.; van Gils, J.M.; Beckers, C.; Horrevoets, A.J.; Hordijk, P.L.; Zwaginga, J.-J. P-Selectin Glycoprotein Ligand-1 Is Expressed on Endothelial Cells and Mediates Monocyte Adhesion to Activated Endothelium. Arter. Thromb. Vasc. Biol. 2007, 27, 1023–1029.
- Gong, L.; Cai, Y.; Zhou, X.; Yang, H. Activated Platelets Interact with Lung Cancer Cells Through P-Selectin Glycoprotein Ligand-1. Pathol. Oncol. Res. 2012, 18, 989–996.
- Sang, Y.; Roest, M.; de Laat, B.; de Groot, P.G.; Huskens, D. Interplay between platelets and coagulation. Blood Rev. 2020, 46, 100733.
- Yan, M.; Lesyk, G.; Radziwon-Balicka, A.; Jurasz, P. Pharmacological Regulation of Platelet Factors That Influence Tumor Angiogenesis. Semin. Oncol. 2014, 41, 370–377.
- Raica, M.; Cimpean, A.M. Platelet-Derived Growth Factor (PDGF)/PDGF Receptors (PDGFR) Axis as Target for Antitumor and Antiangiogenic Therapy. Pharmaceuticals 2010, 3, 572–599.
- Langer, H.; May, A.E.; Daub, K.; Heinzmann, U.; Lang, P.; Schumm, M.; Vestweber, D.; Massberg, S.; Schönberger, T.; Pfisterer, I.; et al. Adherent Platelets Recruit and Induce Differentiation of Murine Embryonic Endothelial Progenitor Cells to Mature Endothelial Cells In Vitro. Circ. Res. 2006, 98, e2–e10.
- Trikha, M.; Zhou, Z.; Timar, J.; Raso, E.; Kennel, M.; Emmell, E.; Nakada, M.T. Multiple roles for platelet GPIIb/IIIa and alphavbeta3 integrins in tumor growth, angiogenesis, and metastasis. Cancer Res. 2002, 62, 2824–2833.
- Ho-Tin-Noé, B.; Goerge, T.; Cifuni, S.M.; Duerschmied, D.; Wagner, D.D. Platelet Granule Secretion Continuously Prevents Intratumor Hemorrhage. Cancer Res. 2008, 68, 6851–6858.
- Schumacher, D.; Strilic, B.; Sivaraj, K.K.; Wettschureck, N.; Offermanns, S. Platelet-Derived Nucleotides Promote Tumor-Cell Transendothelial Migration and Metastasis via P2Y2 Receptor. Cancer Cell 2013, 24, 130–137.
- Sun, C.; Feng, S.-B.; Cao, Z.-W.; Bei, J.-J.; Chen, Q.; Xu, X.-J.; Zhou, Z.; Yu, Z.-P.; Hu, H.-Y. Up-Regulated Expression of Matrix Metalloproteinases in Endothelial Cells Mediates Platelet Microvesicle-Induced Angiogenesis. Cell. Physiol. Biochem. 2017, 41, 2319–2332.
- Yuan, L.; Liu, X. Platelets are associated with xenograft tumor growth and the clinical malignancy of ovarian cancer through an angiogenesis-dependent mechanism. Mol. Med. Rep. 2014, 11, 2449–2458.
- Artini, P.G.; Ruggiero, M.; Monteleone, P.; Carpi, A.; Cristello, F.; Cela, V.; Genazzani, A.R. Vascular endothelial growth factor and its soluble receptor in benign and malignant ovarian tumors. Biomed. Pharmacother. 2008, 62, 373–377.
- Byrne, A.T.; Ross, L.; Holash, J.; Nakanishi, M.; Hu, L.; Hofmann, J.I.; Yancopoulos, G.D.; Jaffe, R.B. Vascular endothelial growth factor-trap decreases tumor burden, inhibits ascites, and causes dramatic vascular remodeling in an ovarian cancer model. Clin. Cancer Res. 2003, 9, 5721–5728.
- Li, L.; Wang, L.; Zhang, W.; Tang, B.; Zhang, J.; Song, H.; Yao, D.; Tang, Y.; Chen, X.; Yang, Z.; et al. Correlation of serum VEGF levels with clinical stage, therapy efficacy, tumor metastasis and patient survival in ovarian cancer. Anticancer. Res. 2004, 24, 1973–1979.
- Matei, D.; Kelich, S.; Cao, L.; Menning, N.; Emerson, R.E.; Rao, J.; Jeng, M.H.; Sledge, G.W. PDGF BB induces VEGF secretion in ovarian cancer. Cancer Biol. Ther. 2007, 6, 1951–1959.
- Silini, A.; Ghilardi, C.; Figini, S.; Sangalli, F.; Fruscio, R.; Dahse, R.; Pedley, R.B.; Giavazzi, R.; Bani, M. Regulator of G-protein signaling 5 (RGS5) protein: A novel marker of cancer vasculature elicited and sustained by the tumor’s proangiogenic microenvironment. Cell. Mol. Life Sci. 2011, 69, 1167–1178.
- Bergmeier, W.; Piffath, C.L.; Goerge, T.; Cifuni, S.M.; Ruggeri, Z.M.; Ware, J.; Wagner, D.D. The role of platelet adhesion receptor GPIbα far exceeds that of its main ligand, von Willebrand factor, in arterial thrombosis. Proc. Natl. Acad. Sci. USA 2006, 103, 16900–16905.
- Borsig, L.; Wong, R.; Feramisco, J.; Nadeau, D.R.; Varki, N.M.; Varki, A. Heparin and cancer revisited: Mechanistic connections involving platelets, P-selectin, carcinoma mucins, and tumor metastasis. Proc. Natl. Acad. Sci. USA 2001, 98, 3352–3357.
- Zhang, N.; Zhang, W.-J.; Cai, H.-Q.; Liu, H.-L.; Peng, L.; Li, C.-H.; Ye, L.-Y.; Xu, S.-Q.; Yang, Z.-H.; Lou, J.-N. Platelet adhesion and fusion to endothelial cell facilitate the metastasis of tumor cell in hypoxia-reoxygenation condition. Clin. Exp. Metastasis 2010, 28, 1–12.
- Stacker, S.A.; Williams, S.P.; Karnezis, T.; Shayan, R.; Fox, S.B.; Achen, M.G. Lymphangiogenesis and lymphatic vessel remodelling in cancer. Nat. Rev. Cancer 2014, 14, 159–172.
- Suzuki-Inoue, K. Essential in vivo roles of the platelet activation receptor CLEC-2 in tumour metastasis, lymphangiogenesis and thrombus formation. J. Biochem. 2011, 150, 127–132.
- Lim, L.; Bui, H.; Farrelly, O.; Yang, J.; Li, L.; Enis, D.; Ma, W.; Chen, M.; Oliver, G.; Welsh, J.D.; et al. Hemostasis stimulates lymphangiogenesis through release and activation of VEGFC. Blood 2019, 134, 1764–1775.
- Da, M.-X.; Wu, Z.; Tian, H.-W. Tumor Lymphangiogenesis and Lymphangiogenic Growth Factors. Arch. Med. Res. 2008, 39, 365–372.
- Sopo, M.; Anttila, M.; Muukkonen, O.-T.; Ylä-Herttuala, S.; Kosma, V.-M.; Keski-Nisula, L.; Sallinen, H. Microvessels in Epithelial Ovarian Tumors: High Microvessel Density Is a Significant Feature of Malignant Ovarian Tumors. Anticancer. Res. 2020, 40, 6923–6931.
- Kitano, H.; Kageyama, S.-I.; Hewitt, S.M.; Hayashi, R.; Doki, Y.; Ozaki, Y.; Fujino, S.; Takikita, M.; Kubo, H.; Fukuoka, J. Podoplanin Expression in Cancerous Stroma Induces Lymphangiogenesis and Predicts Lymphatic Spread and Patient Survival. Arch. Pathol. Lab. Med. 2010, 134, 1520–1527.
- Bianchi, R.; Fischer, E.; Yuen, D.; Ernst, E.; Ochsenbein, A.M.; Chen, L.; Otto, V.I.; Detmar, M. Mutation of Threonine 34 in Mouse Podoplanin-Fc Reduces CLEC-2 Binding and Toxicity in Vivo While Retaining Anti-lymphangiogenic Activity. J. Biol. Chem. 2014, 289, 21016–21027.
- Zhao, R.-W.; Yang, S.-H.; Cai, L.-Q.; Zhang, J.; Wang, J.; Wang, Z.-H. Roles of vascular endothelial growth factor and platelet-derived growth factor in lymphangiogenesis in epithelial ovarian carcinoma. Zhonghua Fu Chan Ke Za Zhi 2009, 44, 760–764.
- Krishnapriya, S.; Sidhanth, C.; Manasa, P.; Sneha, S.; Bindhya, S.; Nagare, R.P.; Ramachandran, B.; Vishwanathan, P.; Murhekar, K.; Shirley, S.; et al. Cancer stem cells contribute to angiogenesis and lymphangiogenesis in serous adenocarcinoma of the ovary. Angiogenesis 2019, 22, 441–455.
- Liao, S.; Liu, J.; Lin, P.; Shi, T.; Jain, R.K.; Xu, L. TGF-β Blockade Controls Ascites by Preventing Abnormalization of Lymphatic Vessels in Orthotopic Human Ovarian Carcinoma Models. Clin. Cancer Res. 2011, 17, 1415–1424.
- Sweeney, M.; Ayyadurai, S.; Zlokovic, B.V. Pericytes of the neurovascular unit: Key functions and signaling pathways. Nat. Neurosci. 2016, 19, 771–783.
- Xian, X.; Håkansson, J.; Ståhlberg, A.; Lindblom, P.; Betsholtz, C.; Gerhardt, H.; Semb, H. Pericytes limit tumor cell metastasis. J. Clin. Investig. 2006, 116, 642–651.
- Raza, A.; Franklin, M.J.; Dudek, A.Z. Pericytes and vessel maturation during tumor angiogenesis and metastasis. Am. J. Hematol. 2010, 85, 593–598.
- Bergers, G.; Song, S. The role of pericytes in blood-vessel formation and maintenance. Neuro-Oncology 2005, 7, 452–464.
- Sinha, D.; Chong, L.; George, J.; Schlueter, H.; Mönchgesang, S.; Mills, S.; Jason Australian Ovarian Cancer Study Group; Parish, C.R.; Bowtell, D.D.; Kaur, P. Pericytes Promote Malignant Ovarian Cancer Progression in Mice and Predict Poor Prognosis in Serous Ovarian Cancer Patients. Clin. Cancer Res. 2016, 22, 1813–1824.
- Sasano, T.; Gonzalez-Delgado, R.; Muñoz, N.M.; Carlos-Alcade, W.; Cho, M.S.; Sheth, R.A.; Sood, A.K.; Afshar-Kharghan, V. Podoplanin promotes tumor growth, platelet aggregation, and venous thrombosis in murine models of ovarian cancer. J. Thromb. Haemost. 2021, 20, 104–114.
- Armulik, A.; Genové, G.; Betsholtz, C. Pericytes: Developmental, Physiological, and Pathological Perspectives, Problems, and Promises. Dev. Cell 2011, 21, 193–215.
- Zonneville, J.; Safina, A.; Truskinovsky, A.M.; Arteaga, C.L.; Bakin, A.V. TGF-β signaling promotes tumor vasculature by enhancing the pericyte-endothelium association. BMC Cancer 2018, 18, 670.
- Zhang, L.; Yang, N.; Park, J.-W.; Katsaros, D.; Fracchioli, S.; Cao, G.; O’Brien-Jenkins, A.; Randall, T.C.; Rubin, S.C.; Coukos, G. Tumor-derived vascular endothelial growth factor up-regulates angiopoietin-2 in host endothelium and destabilizes host vasculature, supporting angiogenesis in ovarian cancer. Cancer Res. 2003, 63, 3403–3412.
- Sallinen, H.; Anttila, M.; Gröhn, O.; Koponen, J.; Hämäläinen, K.; Kholova, I.; Kosma, V.-M.; Heinonen, S.; Alitalo, K.; Ylä-Herttuala, S. Cotargeting of VEGFR-1 and -3 and angiopoietin receptor Tie2 reduces the growth of solid human ovarian cancer in mice. Cancer Gene Ther. 2010, 18, 100–109.
- Heldin, C.-H.; Westermark, B. Mechanism of Action and In Vivo Role of Platelet-Derived Growth Factor. Physiol. Rev. 1999, 79, 1283–1316.
- Lu, C.; Shahzad, M.M.; Moreno-Smith, M.; Lin, Y.; Jennings, N.B.; Allen, J.K.; Landen, C.N.; Mangala, L.S.; Armaiz-Pena, G.N.; Schmandt, R.; et al. Targeting pericytes with a PDGF-B aptamer in human ovarian carcinoma models. Cancer Biol. Ther. 2010, 9, 176–182.
- Taraboletti, G.; Roberts, D.; Liotta, L.A.; Giavazzi, R. Platelet thrombospondin modulates endothelial cell adhesion, motility, and growth: A potential angiogenesis regulatory factor. J. Cell Biol. 1990, 111, 765–772.
- Bagavandoss, P.; Wilks, J. Specific inhibition of endothelial cell proliferation by thrombospondin. Biochem. Biophys. Res. Commun. 1990, 170, 867–872.
- Matuszewska, K.; Kortenaar, S.T.; Pereira, M.; Santry, L.A.; Petrik, D.; Lo, K.-M.; Bridle, B.W.; Wootton, S.K.; Lawler, J.; Petrik, J. Addition of an Fc-IgG induces receptor clustering and increases the in vitro efficacy and in vivo anti-tumor properties of the thrombospondin-1 type I repeats (3TSR) in a mouse model of advanced stage ovarian cancer. Gynecol. Oncol. 2021, 164, 154–169.
- Russell, S.; Duquette, M.; Liu, J.; Drapkin, R.; Lawler, J.; Petrik, J. Combined therapy with thrombospondin-1 type I repeats (3TSR) and chemotherapy induces regression and significandy improves survival in a preclinical model of advanced stage epithelial ovarian cancer. FASEB J. 2014, 29, 576–588.
- Matuszewska, K.; Santry, L.A.; van Vloten, J.P.; Auyeung, A.W.K.; Major, P.P.; Lawler, J.; Wootton, S.K.; Bridle, B.W.; Petrik, J. Combining Vascular Normalization with an Oncolytic Virus Enhances Immunotherapy in a Preclinical Model of Advanced-Stage Ovarian Cancer. Clin. Cancer Res. 2019, 25, 1624–1638.
- Boucharaba, A.; Serre, C.-M.; Grès, S.; Saulnier-Blache, J.S.; Bordet, J.-C.; Guglielmi, J.; Clézardin, P.; Peyruchaud, O. Platelet-derived lysophosphatidic acid supports the progression of osteolytic bone metastases in breast cancer. J. Clin. Investig. 2004, 114, 1714–1725.
- Gopinathan, G.; Milagre, C.; Pearce, O.M.; Reynolds, L.E.; Hodivala-Dilke, K.; Leinster, D.A.; Zhong, H.; Hollingsworth, R.E.; Thompson, R.; Whiteford, J.R.; et al. Interleukin-6 Stimulates Defective Angiogenesis. Cancer Res. 2015, 75, 3098–3107.
- Merritt, W.M.; Nick, A.M.; Carroll, A.R.; Lu, C.; Matsuo, K.; Dumble, M.; Jennings, N.; Zhang, S.; Lin, Y.G.; Spannuth, W.A.; et al. Bridging the Gap between Cytotoxic and Biologic Therapy with Metronomic Topotecan and Pazopanib in Ovarian Cancer. Mol. Cancer Ther. 2010, 9, 985–995.
- Tullemans, B.M.E.; Nagy, M.; Sabrkhany, S.; Griffioen, A.W.; Egbrink, M.G.A.O.; Aarts, M.; Heemskerk, J.W.M.; Kuijpers, M.J.E. Tyrosine Kinase Inhibitor Pazopanib Inhibits Platelet Procoagulant Activity in Renal Cell Carcinoma Patients. Front. Cardiovasc. Med. 2018, 5, 142.
- Tracy, L.E.; Minasian, R.A.; Caterson, E. Extracellular Matrix and Dermal Fibroblast Function in the Healing Wound. Adv. Wound Care 2016, 5, 119–136.
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186.
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598.
- Luga, V.; Zhang, L.; Viloria-Petit, A.M.; Ogunjimi, A.A.; Inanlou, M.R.; Chiu, E.; Buchanan, M.; Hosein, A.N.; Basik, M.; Wrana, J.L. Exosomes Mediate Stromal Mobilization of Autocrine Wnt-PCP Signaling in Breast Cancer Cell Migration. Cell 2012, 151, 1542–1556.
- Hu, Y.; Yan, C.; Mu, L.; Huang, K.; Li, X.; Tao, D.; Wu, Y.; Qin, J. Fibroblast-derived exosomes contribute to chemoresistance through priming cancer stem cells in colorectal cancer. PLoS ONE 2015, 10, e0125625.
- Ronca, R.; van Ginderachter, J.; Turtoi, A. Paracrine interactions of cancer-associated fibroblasts, macrophages and endothelial cells: Tumor allies and foes. Curr. Opin. Oncol. 2018, 30, 45–53.
- Kojima, Y.; Acar, A.; Eaton, E.N.; Mellody, K.T.; Scheel, C.; Ben-Porath, I.; Onder, T.T.; Wang, Z.C.; Richardson, A.L.; Weinberg, R.A.; et al. Autocrine TGF-β and stromal cell-derived factor-1 (SDF-1) signaling drives the evolution of tumor-promoting mammary stromal myofibroblasts. Proc. Natl. Acad. Sci. USA 2010, 107, 20009–20014.
- Anderberg, C.; Li, H.; Fredriksson, L.; Andrae, J.; Betsholtz, C.; Li, X.; Eriksson, U.; Pietras, K. Paracrine Signaling by Platelet-Derived Growth Factor-CC Promotes Tumor Growth by Recruitment of Cancer-Associated Fibroblasts. Cancer Res. 2009, 69, 369–378.
- Aoto, K.; Ito, K.; Aoki, S. Complex formation between platelet-derived growth factor receptor β and transforming growth factor β receptor regulates the differentiation of mesenchymal stem cells into cancer-associated fibroblasts. Oncotarget 2018, 9, 34090–34102.
- Zeltz, C.; Alam, J.; Liu, H.; Erusappan, P.M.; Hoschuetzky, H.; Molven, A.; Parajuli, H.; Cukierman, E.; Costea, D.-E.; Lu, N.; et al. α11β1 Integrin is Induced in a Subset of Cancer-Associated Fibroblasts in Desmoplastic Tumor Stroma and Mediates In Vitro Cell Migration. Cancers 2019, 11, 765.
- Fukagawa, D.; Sugai, T.; Osakabe, M.; Suga, Y.; Nagasawa, T.; Itamochi, H.; Sugiyama, T. Protein expression patterns in cancer-associated fibroblasts and cells undergoing the epithelial-mesenchymal transition in ovarian cancers. Oncotarget 2018, 9, 27514–27524.
- Radhakrishnan, R.; Ha, J.H.; Jayaraman, M.; Liu, J.; Moxley, K.M.; Isidoro, C.; Sood, A.K.; Song, Y.S.; Dhanasekaran, D.N. Ovarian cancer cell-derived lysophosphatidic acid induces glycolytic shift and cancer-associated fibroblast-phenotype in normal and peritumoral fibroblasts. Cancer Lett. 2018, 442, 464–474.
- Rother, E.; Brandl, R.; Baker, D.L.; Goyal, P.; Gebhard, H.; Tigyi, G.; Siess, W. Subtype-Selective Antagonists of Lysophosphatidic Acid Receptors Inhibit Platelet Activation Triggered by the Lipid Core of Atherosclerotic Plaques. Circulation 2003, 108, 741–747.
- Jeon, E.S.; Heo, S.C.; Lee, I.H.; Choi, Y.J.; Park, J.H.; Choi, K.U.; Park, Y.; Suh, D.-S.; Yoon, M.-S.; Kim, J.H. Ovarian cancer-derived lysophosphatidic acid stimulates secretion of VEGF and stromal cell-derived factor-1α from human mesenchymal stem cells. Exp. Mol. Med. 2010, 42, 280–293.
- Chignard, M.; Le Couedic, J.P.; Tence, M.; Vargaftig, B.B.; Benveniste, J. The role of platelet-activating factor in platelet aggregation. Nature 1979, 279, 799–800.
- Cho, J.A.; Park, H.; Lim, E.H.; Kim, K.H.; Choi, J.S.; Lee, J.H.; Shin, J.W.; Lee, K.W. Exosomes from ovarian cancer cells induce adipose tissue-derived mesenchymal stem cells to acquire the physical and functional characteristics of tumor-supporting myofibroblasts. Gynecol. Oncol. 2011, 123, 379–386.
- So, K.A.; Min, K.J.; Hong, J.H.; Lee, J.-K. Interleukin-6 expression by interactions between gynecologic cancer cells and human mesenchymal stem cells promotes epithelial-mesenchymal transition. Int. J. Oncol. 2015, 47, 1451–1459.
- Gastl, G.; Plante, M.; Finstad, C.L.; Wong, G.Y.; Federici, M.G.; Bander, N.H.; Rubin, S.C. High IL-6 levels in ascitic fluid correlate with reactive thrombocytosis in patients with epithelial ovarian cancer. Br. J. Haematol. 1993, 83, 433–441.
- Shield, K.; Ackland, M.L.; Ahmed, N.; Rice, G.E. Multicellular spheroids in ovarian cancer metastases: Biology and pathology. Gynecol. Oncol. 2009, 113, 143–148.
- Rynne-Vidal, A.; Au-Yeung, C.L.; Jiménez-Heffernan, J.A.; Perez-Lozano, M.-L.; Cremades-Jimeno, L.; Bárcena, C.; Cristobal, I.; Fernández-Chacón, C.; Yeung, T.L.; Mok, S.C.; et al. Mesothelial-to-mesenchymal transition as a possible therapeutic target in peritoneal metastasis of ovarian cancer. J. Pathol. 2017, 242, 140–151.
- Matte, I.; Lane, D.; Laplante, C.; Rancourt, C.; Piché, A. Profiling of cytokines in human epithelial ovarian cancer ascites. Am. J. Cancer Res. 2012, 2, 566–580.
- Yan, D.; Liu, X.; Xu, H.; Guo, S.-W. Mesothelial Cells Participate in Endometriosis Fibrogenesis through Platelet-Induced Mesothelial-Mesenchymal Transition. J. Clin. Endocrinol. Metab. 2020, 105, e4124–e4147.
- Steidel, C.; Ender, F.; Rody, A.; von Bubnoff, N.; Gieseler, F. Biologically Active Tissue Factor-Bearing Larger Ectosome-Like Extracellular Vesicles in Malignant Effusions from Ovarian Cancer Patients: Correlation with Incidence of Thrombosis. Int. J. Mol. Sci. 2021, 22, 790.
- Orellana, R.; Kato, S.; Erices, R.; Bravo, M.L.; Gonzalez, P.; Oliva, B.; Cubillos, S.; Valdivia, A.; Ibañez, C.; Brañes, J.; et al. Platelets enhance tissue factor protein and metastasis initiating cell markers, and act as chemoattractants increasing the migration of ovarian cancer cells. BMC Cancer 2015, 15, 290.
- Malacrida, B.; Nichols, S.; Maniati, E.; Jones, R.; Delanie-Smith, R.; Roozitalab, R.; Tyler, E.J.; Thomas, M.; Boot, G.; Mackerodt, J.; et al. A human multi-cellular model shows how platelets drive production of diseased extracellular matrix and tissue invasion. Iscience 2021, 24, 102676.
- Motohara, T.; Masuda, K.; Morotti, M.; Zheng, Y.; El-Sahhar, S.; Chong, K.Y.; Wietek, N.; Alsaadi, A.; Carrami, E.M.; Hu, Z.; et al. An evolving story of the metastatic voyage of ovarian cancer cells: Cellular and molecular orchestration of the adipose-rich metastatic microenvironment. Oncogene 2018, 38, 2885–2898.
- Ryner, L.; Guan, Y.; Firestein, R.; Xiao, Y.; Choi, Y.; Rabe, C.; Lu, S.; Fuentes, E.; Huw, L.-Y.; Lackner, M.R.; et al. Upregulation of Periostin and Reactive Stroma Is Associated with Primary Chemoresistance and Predicts Clinical Outcomes in Epithelial Ovarian Cancer. Clin. Cancer Res. 2015, 21, 2941–2951.
- Moran-Jones, K.; Gloss, B.S.; Murali, R.; Chang, D.K.; Colvin, E.K.; Jones, M.D.; Yuen, S.; Howell, V.M.; Brown, L.M.; Wong, C.W.; et al. Connective tissue growth factor as a novel therapeutic target in high grade serous ovarian cancer. Oncotarget 2015, 6, 44551–44562.
- Yeung, T.-L.; Leung, C.S.; Wong, K.-K.; Samimi, G.; Thompson, M.S.; Liu, J.; Zaid, T.M.; Ghosh, S.; Birrer, M.J.; Mok, S.C. TGF-β Modulates Ovarian Cancer Invasion by Upregulating CAF-Derived Versican in the Tumor Microenvironment. Cancer Res. 2013, 73, 5016–5028.
- Ghoneum, A.; Afify, H.; Salih, Z.; Kelly, M.; Said, N. Role of tumor microenvironment in ovarian cancer pathobiology. Oncotarget 2018, 9, 22832–22849.
- Crawford, Y.; Kasman, I.; Yu, L.; Zhong, C.; Wu, X.; Modrusan, Z.; Kaminker, J.; Ferrara, N. PDGF-C Mediates the Angiogenic and Tumorigenic Properties of Fibroblasts Associated with Tumors Refractory to Anti-VEGF Treatment. Cancer Cell 2009, 15, 21–34.
- Wawro, M.E.; Sobierajska, K.; Ciszewski, W.M.; Niewiarowska, J. Nonsteroidal Anti-Inflammatory Drugs Prevent Vincristine-Dependent Cancer-Associated Fibroblasts Formation. Int. J. Mol. Sci. 2019, 20, 1941.