Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Camila Xu and Version 1 by Alessandro Paiardini.
c-Myc and the other protein family members (i.e., N-Myc and L-Myc), collectively known as “Myc”, are ubiquitous basic helix–loop–helix–leucine zipper (bHLH-LZ) transcription factors that are critical for several cellular processes during cancer genesis and progression. The importance of kinases in Myc regulation goes beyond their ability to phosphorylate the protein. Some kinases can also indirectly affect Myc protein stability by inducing the degradation of the ubiquitin ligase (PLK1 and PKA). Additionally, some kinases physically interact with Myc, protecting it from proteasomal degradation, such as Aurora-A in neuroblastoma.
- Myc
- kinases
- PLK1
- Aurora-A
- Aurora-B
- GSK-3
1. Introduction
c-Myc and the other protein family members (i.e., N-Myc and L-Myc), collectively known as “Myc”, are ubiquitous basic helix–loop–helix–leucine zipper (bHLH-LZ) transcription factors that are critical for several cellular processes during cancer genesis and progression [1]. Indeed, Myc plays a central role among the molecular factors that drive tumour progression. c-Myc was first described as a viral oncoprotein able to induce myelocytomatosis in chickens after retroviral infection, and soon its derivation from a highly conserved vertebrate cellular gene was demonstrated [2,3][2][3]. The other members of the Myc family were subsequently discovered: N-Myc in neuroblastoma [4,5][4][5] and L-Myc in small cell lung carcinoma [6]. Since then, the Myc family members have been widely recognized as potential oncogenes and have been thoroughly studied for their structure, function, and regulation (as described in more detail by others [7,8][7][8]). Decades after its discovery, Myc continues to impress with its involvement in diverse pathways. The complexity of the Myc protein interaction network is reflected in the great heterogeneity of their transcriptional program and biomolecular activities [7,8,9][7][8][9].
Upon forming an obligate heterodimer with its protein partner Max, Myc binds to the consensus DNA sequence 5′-CACGTG-3′ (known as the E-box) and drives the expression of its target genes [10] (Figure 1A). As a mild transcriptional modulator, the effects of Myc are amplified through interactions with a wide range of cofactors, co-activators, and chromatin remodelling components [11]. Relevant functional categories of MYC-induced genes include cell cycle control, DNA replication, cell growth and adhesion, cellular bioenergetics (e.g., glycolysis and mitochondrial biogenesis), and anabolic metabolism (e.g., synthesis of amino acids, nucleotides, and lipids) [1,12,13][1][12][13]. In addition, Myc and HIF1 actively induce the expression of glucose transporters and glycolytic enzymes [14], and the former acts as a downstream effector of several signalling cascades: for instance, aberrant Wnt/β-catenin signalling leads to the constitutive, high transcription of the MYC gene [15,16][15][16]; similarly, potent induction of MYC transcription occurs in response to perturbations of the Sonic hedgehog [17], Notch [18[18][19],19], and Janus kinase (JAK)— signal transducer and activator of transcription 3 (STAT3) pathways [20,21][20][21].
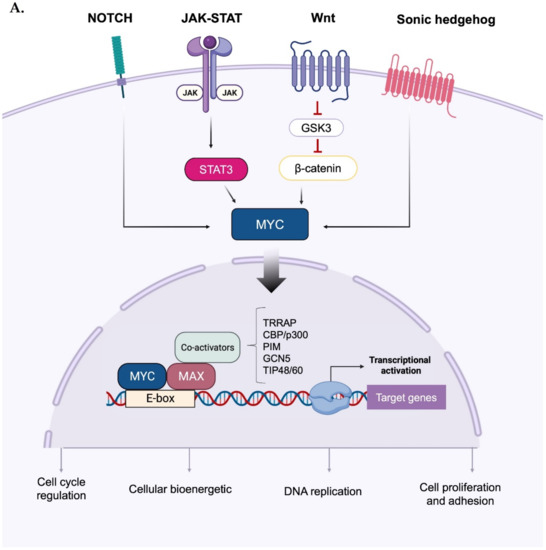
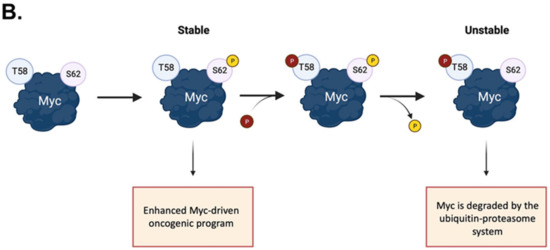
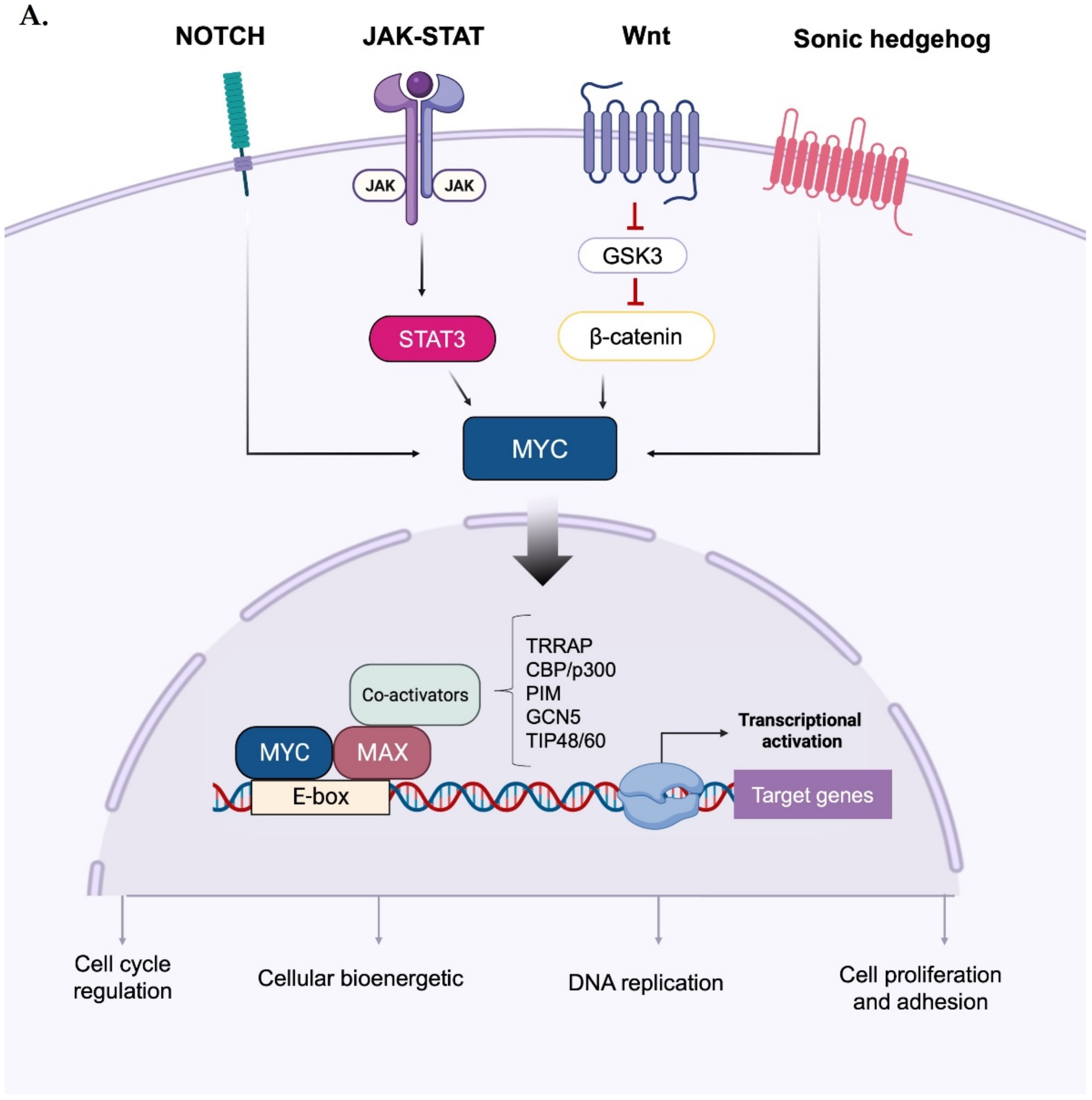
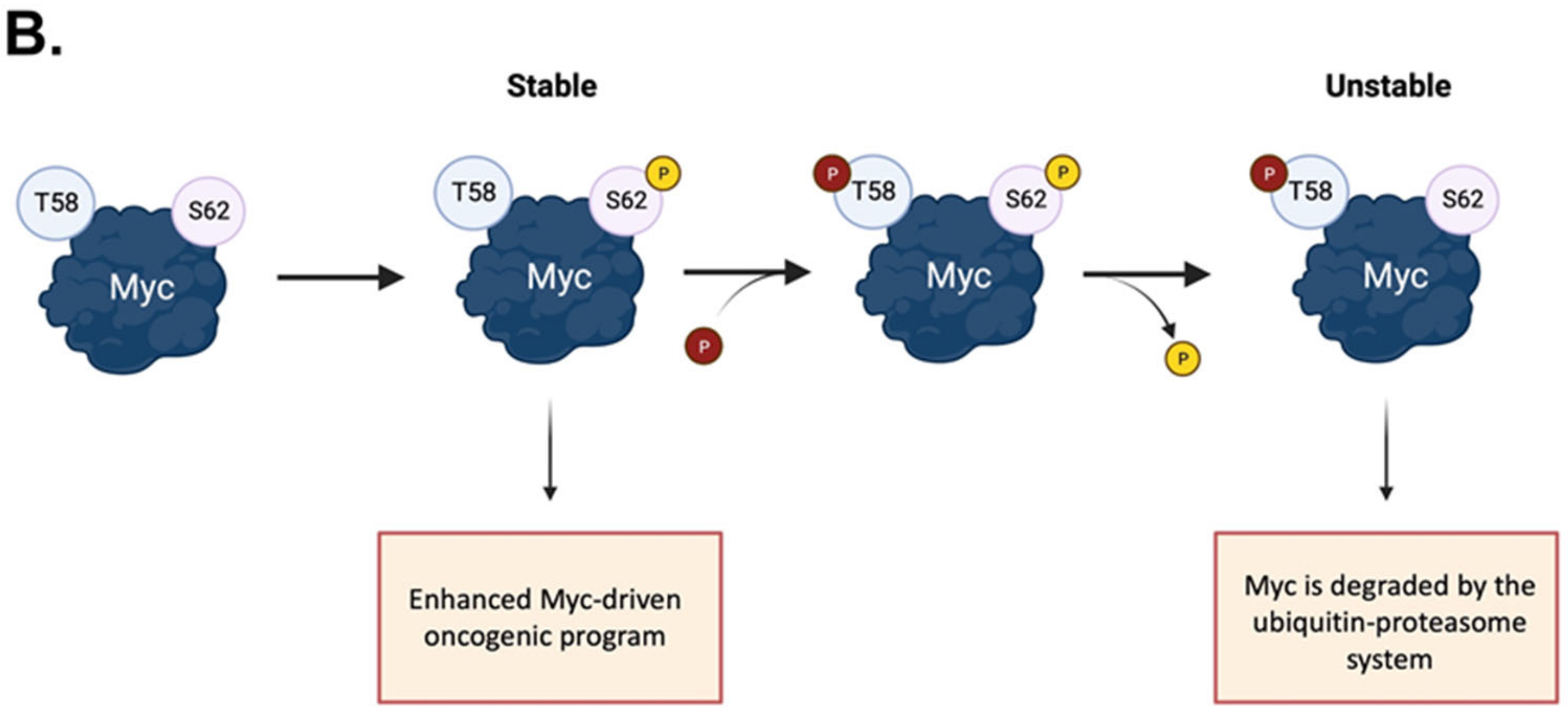
Figure 1. Activation and phospho-dependent stabilization of Myc. (A) Myc can be activated in response to different stimuli via several transduction pathways converging to Myc stabilization. Stabilized Myc associates with its protein partner MAX to form a heterodimer, which, together with other co-activators, binds to the E-box elements and drives the transcription of a target gene subset involved in a wide range of cellular processes. (B) Phospho-dependent stabilization of Myc. Myc displays two key phosphorylation sites that undergo hierarchical phosphorylation, supervising protein stability. Phosphorylation of Myc at the S62 residue determines protein stabilization and primes the subsequent phosphorylation at T58, which induces the removal of the phosphate group at S62; the unstable, singly phosphorylated T58-Myc is then recognized by the ubiquitin ligase Fbxw7 and degraded by the ubiquitin–proteasome system. Created with BioRender.com (accessed on 20 January 2023).
The pleiotropic activities of Myc make its tight regulation mandatory in healthy cells, which need to rapidly turnover Myc proteins. However, steady Myc levels may be affected by pathways that are disrupted in cancer, leading to its enhanced expression and increased stability [22]. Myc mRNA levels are regulated by both transcription initiation (which starts from different promoters [23]) and elongation [24], yielding a transcript that is highly unstable, with a half-life of 20–30 min [25]. Additionally, Myc is rapidly degraded after its synthesis, with a turnover of 25 min [26]. Posttranslational modifications, i.e., acetylation and methylation, contribute to regulating Myc activity and stability [27]. SUMOylation, consisting of the addition of ubiquitin-like molecules to the target protein, is another intriguing observed modification in Myc. The role of the SUMOylation in regulating Myc functions was debated; Myc is SUMOylated in more than 10 lysine residues [28,29,30][28][29][30], but the mutation of all these residues did not abolish Myc SUMOylation, nor alter its activity [28,29,30][28][29][30]. In a particular case, an increased Myc transcription activity was observed: in B cell lymphoma, the SUMOylation mediated by the E3 ligase PIAS1 allows the recognition and phosphorylation of Myc in S62 by JNK1, which precludes the oncoprotein degradation [31]. Noteworthy, in this tumour, a Myc-dependent transcriptional activation of SUMO cascade enzymes was observed. The overall SUMOylation induces G2/M arrest and consequent mitotic abnormalities, which are attributed to the Aurora kinases’ SUMOylation [32]. SUMOylation could also have a role in Myc protein stability, since the inhibition of the proteasome has been shown to increase the levels of SUMOylated Myc [28,33][28][33]. Moreover, the SUMO-specific protease SENP1 has been described to de-SUMOylate Myc, stabilizing the protein [33]. It is possible that the de-SUMOylation drives the subsequent de-ubiquitination on the same residues, since SUMO and ubiquitin modifications can exist on the same lysine residues [34].
The phosphorylation and dephosphorylation of key residues provide the main dynamic regulation of Myc cellular functions: its phosphorylation plays a key role in promoting Myc binding to DNA and in inducing its degradation. The conserved region of Myc called the Myc Box (MB) I is mainly involved in its ubiquitination and degradation [35] (Figure 1B). It contains the residues S62 and T58, which undergo cascade phosphorylation, regulating protein stability and functionality. S62 is a target for a variety of kinases, including ERK, CDKs, and JNK [36], and its phosphorylation correlates with the recruitment of Myc to specific promoters in response to oxidative stress. The S62A mutation has been shown to significantly alter an array of genes involved in apoptosis, proliferation, cellular bioenergetic, and signalling pathways [37]. The phosphorylation of S62 recruits the GSK-3β kinase, which, in turn, phosphorylates T58 on Myc, ensuring its recognition by the Fbxw7 ubiquitin ligase for degradation [38]. T58 and S62 are hotspots of mutation in lymphomas [39,40][39][40] and solid tumours [41,42][41][42]. The non-phosphorylatable T58A mutation leads to the accumulation of pS62 Myc, which presumably repulses Fbxw7 and increases Myc half-life to up to 120 min [40]. Additionally, Myc phosphorylated on T58 reduces the expression of BIM, which is particularly relevant as a tumour suppressor in MYC-driven B cell leukaemia [43]. The phosphorylation of other highly conserved sites, T358, S373, and T400, by the p21-activated kinase PAK2 has been reported to negatively impact on the Myc transcriptional program by interfering with the formation of the Myc–Max–DNA ternary complex [44,45][44][45].
In addition to MBI, other conserved motifs control Myc functions. MBII allows interaction with partners required for transcriptional regulation [46], while MBIII is important for cellular transformation [47]. Finally, MBIV overlaps with the nuclear localization signal [48]. Each motif is conserved among species and ensures the binding of Myc proteins with different partners. Interestingly, the MBI and the MBII motifs overlap with the transactivation domain (TAD), highlighting a well-established correlation between Myc protein stability and transcriptional activity [49].
The importance of kinases in Myc regulation goes beyond their ability to phosphorylate the protein. Some kinases can also indirectly affect Myc protein stability by inducing the degradation of the ubiquitin ligase (PLK1 and PKA, [50,51][50][51]). Additionally, some kinases physically interact with Myc, protecting it from proteasomal degradation, such as Aurora-A in neuroblastoma [52]. The cell-cycle-dependent Aurora-A and Aurora-B form a circuit with Myc, in which Aurora primes the oncogenic program of Myc and vice versa [53,54][53][54]. Aurora-A is also involved in Myc modulation in PKA-dependent tumours [55]. Other kinases are crucial for Myc transcriptional activity (i.e., PIM and BRD4 [56,57,58][56][57][58]). At the same time, Myc enhances the expression of protein kinases, creating a positive feedback loop.
2. Direct and Indirect Myc Regulation by Mitotic Kinases
2.1. PLK1 Kinase
PLK1 is a member of the Polo-like kinases (PLKs) family, which consists of five paralogues: PLK1, PLK2, PLK3, PLK4, and PLK5. It is a conserved serine/threonine kinase that is critical for the proper execution of mitotic events and the maintenance of genome stability during cell division [61,62][59][60]. The overexpression of PLK1 is a hallmark of many types of human cancers, including melanoma, ovarian carcinoma, breast, prostate, thyroid cancers, and glioma [63[61][62][63],64,65], and it is associated with chemoresistance and poor patient outcomes. Genetic ablation or inhibition of PLK1 has been found to affect cell cycle progression, leading to reduced proliferation, apoptosis, and increased sensitivity to chemo- and radiotherapy [66,67,68][64][65][66]. From a structural perspective, PLK1 contains an N-terminal catalytic kinase domain (KD) and a C-terminal region with two Polo-box domains (PBDI and PBDII), which are involved in the phospho-dependent recognition of substrates and are essential for determining the kinase’s spatiotemporal subcellular localization [69][67]. In interphase, the KD and PBD establish an inhibitory interaction that suppresses PLK1 catalytic activity. During mitosis, the PBD provides a docking site for proteins with specific phosphorylated motifs [70][68] and undergoes conformational changes that relieve the inhibition of the KD. This allows the activation loop to be accessed by upstream kinases (such as Aurora-A and its cofactor Bora) for the phosphorylation of the key residue T210, which is necessary for the full functionality of PLK1 [71,72][69][70]. In addition to modulating the kinase activity, the phosphorylation status of PLK1 controls its nuclear translocation and its ubiquitin-mediated degradation, which helps to preserve chromosome segregation fidelity and genome integrity [73,74][71][72]. PLK1 has a complex interactome network involving various pathways beyond mitosis, such as immune response [75][73], epithelial-to-mesenchymal transition [76[74][75],77], and cell death signalling [78,79][76][77]. MYC-amplified tumours often have upregulated PLK1, which creates a feed-forward interaction with Myc that maintains high levels of both proteins, predicting poor prognosis [80,81,82][78][79][80]. The main regulatory role of PLK1 is to enhance Myc stability, as seen in the reduced expression of c-Myc upon PLK1 depletion [83][81]. PLK1 kinase activity is necessary for Myc protein accumulation, through phosphorylation of the key residue S62 [84][82]. In addition, following the phosphorylation of Myc at S279 by PKA [85][83], PLK1 can phosphorylate c-Myc at S281, allowing the ubiquitin ligase SCFβ-TrCP to bind and ubiquitinylate the oncoprotein. This alternative post-translational event increases c-Myc stability and contributes to Myc-dependent cell transformation [51]. However, the main mechanism of Myc stabilization does not seem to involve the phosphorylation of Myc at S62, but rather PLK1′s ability to affect the activity of Myc-degrading machinery. Evidence from the rescue of N-Myc levels in response to PLK1 pharmacological inhibition by using the proteasome inhibitor MG132 led Xiao and colleagues to propose a possible crosstalk between PLK1 and the E3 ubiquitin ligase Fbxw7; their study showed that wild-type PLK1, but not the kinase mutant K82R, suppresses Fbxw7 activity by promoting its phosphorylation and autocatalytic poly-ubiquitination in MYCN-amplified cellular models [50]. A similar PLK1/Fbxw7/Myc axis was also identified in c-MYC-driven medulloblastoma, with PLK1 antagonizing Fbxw7-mediated c-Myc degradation [86][84]. Myc stabilization by PLK1 also plays a prominent role in maintaining the autophagy pathway in tumour cells: knockdown of PLK1 leads to a significant reduction in c-Myc protein levels, impacting on MYC transactivation and impairing Myc-mediated autophagy in osteosarcoma cells [87][85]. While PLK1 modulates Myc stability, the main contribution of Myc to the oncogenic relationship with PLK1 is through transcriptional regulation. This is supported by the presence of a Myc E-box binding site upstream of the PLK1 transcription start site, and the finding that the suppression of c-Myc leads to a corresponding reduction in both PLK1 mRNA and phospho-T210 protein levels in B lymphoma cells expressing a tetracycline-repressible MYC transgene [50,81][50][79]. ChIP experiments performed on neuroblastoma and lymphoma cell lines also demonstrated a significant recruitment of c-Myc to the PLK1 E-box, suggesting that c-Myc directly regulates the PLK1 transcriptional program [50,81][50][79]. Based on these findings, targeting PLK1 has the potential to indirectly target Myc-dependent pathways and address the current challenge of developing a therapeutic approach directed against the undruggable Myc proteins.2.2. Aurora-A and Aurora-B Kinases
An intriguing feedback loop with Myc is described for the serine/threonine Aurora kinases. Three members of this family are present in eukaryotes: Aurora-A and Aurora-B, which are involved in the correct execution of mitosis, and Aurora-C, which is mostly implicated in meiosis [88][86]. Aurora-A and Aurora-B share 71% identity in their kinase domains [89][87] and have complementary functions in mitotic cells. The Aurora-A protein localizes at centrosomes, where it participates in the maturation and separation processes in the G2 and M phases, facilitating the recruitment of PLK1 onto CEP192 [90][88]. During mitosis, it interacts with its major activator, TPX2, allowing for the correct formation and orientation of the bipolar spindle [91,92][89][90]. On the other hand, Aurora-B interacts with INCENP, constituting the chromosome passenger complex (CPC) with Survivin and Borealin, which ensures chromosome cohesion, the correct attachment of microtubules to kinetochores, and cytokinesis [93,94][91][92]. Both proteins are linked to cancer [95,96][93][94]. Aurora-A is frequently overexpressed or amplified in a variety of solid and hematologic (blood-related) tumours [89[87][95],97], and according to the Cancer Genome Atlas (TCGA), it is overexpressed in almost 88% of the tumours observed [98][96]. Aurora-A can promote cell transformation when subjected to a suitable cellular background [99][97]. Additionally, the kinase promotes epithelial-to-mesenchymal transition [100[98][99],101], the expression of self-renewal genes in cancer stem cells [102][100], and cancer cell survival through the regulation of apoptotic modulators (reviewed by [95][93]). Recently, its oncogenic activities were related to its nuclear localization [54]. On the other hand, the contribution of Aurora-B in cancer development is not well understood, despite the fact that it is upregulated in most aneuploid human tumours [89][87] and is a poor prognosis factor in hepatocellular carcinoma, non-small cell lung carcinoma, and oral squamous cell carcinoma [103,104,105][101][102][103]. The oncogenic activities of the Aurora kinases have frequently been linked to Myc proteins. In fact, Myc-driven cancers are susceptible to Aurora kinase deprivation or inhibition. One example is c-Myc-driven B-cell lymphoma, in which the overexpression of both Aurora-A and Aurora-B sensitizes cells to pan-Aurora kinase inhibitor treatment [106][104]. In hepatocellular carcinoma (HCC) cells, Aurora-A interacts with and stabilizes c-Myc, promoting tumour cell survival [107][105]. In neuroendocrine prostate cancer, Aurora-A and N-Myc interact with each other and drive an oncogenic gene expression program [108][106]. In neuroblastoma, Aurora-A depletion mimics the effect of N-Myc deprivation in MYCN-amplified (MNA) but not non-MNA cells. This is due to the stabilizing effect of Aurora-A on N-Myc: the kinase binds to the oncoprotein, preventing the Fbxw7 ubiquitin ligase-mediated ubiquitination of Myc [52]. The reason why Aurora kinases’ impairment is an Achilles’ heel of Myc-driven tumours resides both in their importance for cell division, particularly relevant for proliferating cells, and in their ability to regulate Myc transcription and protein stability. Myc-driven expression of Aurora-A and Aurora-B favours mitotic entry and progression, supporting the active proliferation of cancer cells [106,109][104][107]. Moreover, the Aurora-A/N-Myc complex, which is observed more frequently in S phase, has been suggested to prevent the formation of replication/transcription conflicts induced by the oncogene’s over-transcription activity [110][108]. The first physical interaction described between Myc and Aurora family members was the Aurora-A/N-Myc complex in neuroblastoma cells. Depletion of Aurora-A decreases the half-life of N-Myc from 99 min to 55 min in IMR-32 MNA neuroblastoma cells. This Aurora-A function is independent from its kinase activity, since eight different kinase-deficient mutants are able to stabilize the N-Myc oncoprotein [52]. Aurora-A stabilizes N-Myc by protecting it from recognition by the Fbxw7 ubiquitin ligase; moreover, Aurora-A leads to the accumulation of ubiquitinated N-Myc that is non-K48-linked, suggesting that Aurora-A may recruit ubiquitin ligases/deubiquitinases that create a ubiquitinated N-Myc with less degradable ubiquitin chains [52]. Although it is not clear if Aurora-A preferentially interacts with the double-phosphorylated N-Myc (as proposed by [52], whereas [111][109] did not observe differences between the phosphorylated and unphosphorylated forms for the binding to Aurora-A in vitro), the interplay between Aurora-A and Fbxw7 is well described. In particular, the solution of the crystal structure between the catalytic domain of Aurora-A and the 28–89 N-Myc peptide revealed that Aurora-A binds to the oncoprotein through the MB0 and MBI motifs (residues 61–89) [111][109]. In vitro pull-down experiments showed that Aurora-A competes with Fbxw7 for the binding of N-Myc within residues 61–89. Nevertheless, the phospho-degron of N-Myc remains able to be recognized by the ubiquitin ligase; thus, a complex involving Aurora-A, N-Myc, and the Fbxw7 is still formed [111][109]. The region of N-Myc found in the crystal structure is not present in c-Myc [111][109]. Evidence of Aurora-A/c-Myc binding is controversial, since it was reported that no binding between the two proteins occurs in liver and hepatocellular carcinoma cells [112][110], while on the other hand, Aurora-A and the double-phosphorylated c-Myc co-immunoprecipitated in TP53-altered (deleted or mutated) HCC cells [107][105]. However, Aurora-A does play a role in regulating c-Myc expression. It co-immunoprecipitates with the MYCC promoter within the NHE III1 region, which is described to be particularly important for MYCC transcription [112][110]. Aurora-A has also been shown to act as a co-activator of MYCC transcription in breast cancer [54,113][54][111]. Since Aurora-A lacks DNA binding abilities, Zheng and colleagues identified the ribonucleoprotein hnRNP K as the mediator that allows Aurora-A to activate transcription on the MYCC promoter. Depletion of hnRNP K impairs the recruitment of Aurora-A on the MYCC promoter, but not vice versa [54,113][54][111]. The transactivation activity of Aurora-A is independent of its kinase activity, as the administration of VX-680 and MLN8237 kinase inhibitors does not affect the expression of the oncogene, in contrast to what can be observed after Aurora-A depletion [113][111]. hnRNP K also transcriptionally co-activates the p53 protein [114][112], which negatively impacts the expression of several cell cycle genes, including MYCC [115,116][113][114]. Aurora-A may be involved in this process by phosphorylating the ribonucleoprotein at S379, leading to the disruption of its interaction with p53 [115][113]. On the other hand, c-Myc enhances Aurora-A expression. Mouse fibroblasts with overexpressed c-Myc have a two- to three-fold increase in Aurora-A promoter activity [106][104]. Using an inducible Myc-ER construct in ChIP experiments, den Hollander and colleagues found that the mouse Aurora-A gene containing two E-boxes is enriched upon induction of c-Myc, suggesting that the oncoprotein directly binds to the Aurora-A promoter, although they were not able to detect the recruitment of c-Myc on either of the Aurora kinase human genes [106][104]. Lu and colleagues also found a correlation between Aurora-A and c-Myc mRNA in HCC cells, and used ChIP experiments to show that c-Myc binds to the Aurora-A promoter in the highly conserved E-box regions within the CPG islands [112][110]. In light of their roles in modulating Myc expression and stability, it is interesting to highlight the similarity between the two Aurora kinases. For example, the specificity for the binding of Aurora-A to its activator TPX2 is determined by only one residue, i.e., G198, which is different from that in Aurora-B, N142 [117][115]. Moreover, the G198N substitution is sufficient to convert Aurora-A into a kinetochore-localized Aurora-B-like kinase [117,118,119,120][115][116][117][118]. Furthermore, Aurora-A/INCENP binding has also been observed [121][119]. This evidence suggests a sort of interchangeability between Aurora kinases A and B. However, despite the similarity between Aurora-A/Aurora-B and c-Myc/N-Myc, the interplay with each other is quite different. Aurora-A stabilizes the N-Myc protein [52], and, in turn, high levels of N-Myc directly or indirectly increase Aurora-A mRNA levels (e.g., [122][120]), establishing a feedback loop that feeds itself. Conversely, Aurora-B does not affect N-Myc protein levels in neuroblastoma [123,124][121][122] or retinoblastoma cells [125][123] upon depletion. However, Aurora-B levels do decrease after N-Myc knockdown [125][123]. Aurora-B is also transcriptionally activated by direct binding of N-Myc to motifs upstream of the transcription start site [124,125][122][123] and by indirect binding of c-Myc in mice, resulting in a 30-fold increase in Aurora-B promoter activity upon Myc-ER activation [106][104]. It is worth mentioning that Jiang and his colleagues recently identified a novel regulatory mechanism for Aurora B that can stabilize c-Myc through kinase-dependent activity in acute lymphoblastic leukaemia. Experimental results show that Aurora B directly phosphorylates c-Myc at the S67 residue, thereby promoting its stability by counteracting GSK-3-mediated T58 phosphorylation. Notably, sequence alignment did not reveal the S67 phosphorylation site in N-Myc, potentially explaining the non-regulatory role of Aurora B in MYCN-amplified cancers [53]. In conclusion, the Myc and Aurora family of proteins cross-regulate each other, establishing feedback mechanisms that are particularly relevant for cancer progression. In addition, Aurora-A also impacts the expression and stability of Myc proteins by participating in other processes, such as phosphorylating several members of the PI3K/AKT and Wnt/β-catenin pathways, or proteins involved in Myc stability, such as GSK3β and p53 [101,126,127,128][99][124][125][126]. It is clear that untangling this intricate network of interactions that affects both the cell cycle and the activity/stability of Myc will need more in-depth investigation.References
- Scafuro, M.; Capasso, L.; Carafa, V.; Altucci, L.; Nebbioso, A. Gene transactivation and transrepression in myc-driven cancers. Int. J. Mol. Sci. 2021, 22, 3458.
- Sheiness, D.; Bishop, J.M. DNA and RNA from Uninfected Vertebrate Cells Contain Nucleotide Sequences Related to the Putative Transforming Gene of Avian Myelocytomatosis Virus. J. Virol. 1979, 31, 514–521.
- Roussel, M.; Saule, S.; Lagrou, C.; Rommens, C.; Beug, H.; Graf, T.; Stehelin, D. Three new types of viral oncogene of cellular origin specific for haematopoietic cell transformation. Nature 1979, 281, 452–455.
- Schwab, M.; Alitalo, K.; Klempnauer, K.-H.; Varmus, H.E.; Bishop, J.M.; Gilbert, F.; Brodeur, G.; Goldstein, M.; Trent, J. Amplified DNA with limited homology to myc cellular oncogene is shared by human neuroblastoma cell lines and a neuroblastoma tumour. Nature 1983, 305, 245–248.
- Kohl, N.E.; Kanda, N.; Schreck, R.R.; Bruns, G.; Latt, S.A.; Gilbert, F.; Alt, F.W. Transposition and amplification of oncogene-related sequences in human neuroblastomas. Cell 1983, 35 Pt 1, 359–367.
- Nau, M.M.; Brooks, B.J.; Battey, J.F.; Sausville, E.; Gazdar, A.F.; Kirsch, I.R.; McBride, O.W.; Bertness, V.L.; Hollis, G.F.; Minna, J.D. L-myc, a new myc-related gene amplified and expressed in human small cell lung cancer. Nature 1985, 318, 69–73.
- Das, S.K.; Lewis, B.A.; Levens, D. MYC: A complex problem. Trends Cell Biol. 2023, 33, 235–246. Available online: http://www.ncbi.nlm.nih.gov/pubmed/35963793 (accessed on 22 February 2023).
- Lourenco, C.; Resetca, D.; Redel, C.; Lin, P.; MacDonald, A.S.; Ciaccio, R.; Kenney, T.M.G.; Wei, Y.; Andrews, D.W.; Sunnerhagen, M.; et al. MYC protein interactors in gene transcription and cancer. Nat Rev Cancer 2021, 21, 579–591.
- Conacci-Sorrell, M.; McFerrin, L.; Eisenman, R.N. An overview of MYC and its interactome. Cold Spring Harb. Perspect. Med. 2014, 4, a014357.
- Adhikary, S.; Eilers, M. Transcriptional regulation and transformation by Myc proteins. Nat. Rev. Mol. Cell. Biol. 2005, 6, 635–645.
- Hann, S.R. MYC cofactors: Molecular switches controlling diverse biological outcomes. Cold Spring Harb. Perspect. Med. 2014, 4, a014399.
- Hartl, M. The quest for targets executing MYC-dependent cell transformation. Front. Oncol. 2016, 6, 132.
- Tu, W.B.; Helander, S.; Pilstål, R.; Hickman, K.A.; Lourenco, C.; Jurisica, I.; Raught, B.; Wallner, B.; Sunnerhagen, M.; Penn, L.Z. Myc and its interactors take shape. Biochim. Biophys. Acta-Gene Regul. Mech. 2015, 1849, 469–483.
- Santinon, G.; Enzo, E.; Dupont, S. The sweet side of YAP/TAZ. Cell Cycle 2015, 14, 2543–2544.
- Rennoll, S. Regulation of MYC gene expression by aberrant Wnt/β-catenin signaling in colorectal cancer. World J. Biol. Chem. 2015, 6, 290.
- Zhang, S.; Li, Y.; Wu, Y.; Shi, K.; Bing, L.; Hao, J. Wnt/β-Catenin Signaling Pathway Upregulates c-Myc Expression to Promote Cell Proliferation of P19 Teratocarcinoma Cells. Anat. Rec. 2012, 295, 2104–2113.
- Liu, N.; Wang, S.; Cui, Y.; Shen, L.; Du, Y.; Li, G.; Zhang, B.; Wang, R. Sonic hedgehog elevates N-myc gene expression in neural stem cells. Neural Regen. Res. 2012, 7, 1703–1708.
- Palomero, T.; Lim, W.K.; Odom, D.T.; Sulis, M.L.; Real, P.J.; Margolin, A.; Barnes, K.C.; O’Neil, J.; Neuberg, D.; Weng, A.P.; et al. NOTCH1 directly regulates c-MYC and activates a feed-forward-loop transcriptional network promoting leukemic cell growth. Proc. Natl. Acad. Sci. USA 2006, 103, 18261–18266.
- Sanchez-Martin, M.; Ferrando, A. The NOTCH1-MYC highway toward T-cell acute lymphoblastic leukemia. Blood 2017, 129, 1124–1133.
- Huang, L.; Liu, D.; Wang, N.; Ling, S.; Tang, Y.; Wu, J.; Hao, L.; Luo, H.; Hu, X.; Sheng, L.; et al. Integrated genomic analysis identifies deregulated JAK/STAT-MYC-biosynthesis axis in aggressive NK-cell leukemia. Cell Res. 2018, 28, 172–186.
- Jin, W. Role of JAK/STAT3 Signaling in the Regulation of Metastasis, the Transition of Cancer Stem Cells, and Chemoresistance of Cancer by Epithelial–Mesenchymal Transition. Cells 2020, 9, 217.
- Stine, Z.E.; Walton, Z.E.; Altman, B.J.; Hsieh, A.L.; Dang, C.V. MYC, metabolism, and cancer. Cancer Discov. 2015, 5, 1024–1039.
- Battey, J.; Moulding, C.; Taub, R.; Murphy, W.; Stewart, T.; Potter, H.; Lenoir, G.; Leder, P. The human c-myc oncogene: Structural consequences of translocation into the igh locus in Burkitt lymphoma. Cell 1983, 34, 779–787.
- Liu, J.; Levens, D. Making Myc. Curr. Top. Microbiol. Immunol. 2006, 302, 1–32.
- Dani, C.; Blanchard, J.M.; Piechaczyk, M.; El Sabouty, S.; Marty, L.; Jeanteur, P. Extreme instability of myc mRNA in normal and transformed human cells. Proc. Natl. Acad. Sci. USA 1984, 81, 7046–7050.
- Hann, S.R.; Eisenman, R.N. Proteins encoded by the human c-myc oncogene: Differential expression in neoplastic cells. Mol. Cell. Biol. 1984, 4, 2486–2497.
- Farrell, A.S.; Sears, R.C. MYC degradation. Cold Spring Harb. Perspect. Med. 2014, 4, 1–16.
- González-Prieto, R.; Cuijpers, S.A.G.; Kumar, R.; Hendriks, I.A.; Vertegaal, A.C.O. c-Myc is targeted to the proteasome for degradation in a SUMOylation-dependent manner, regulated by PIAS1, SENP7 and RNF4. Cell Cycle 2015, 14, 1859–1872.
- Kalkat, M.; Chan, P.-K.; Wasylishen, A.R.; Srikumar, T.; Kim, S.S.; Ponzielli, R.; Bazett-Jones, D.P.; Raught, B.; Penn, L.Z. Identification of c-MYC SUMOylation by mass spectrometry. PLoS ONE 2014, 9, e115337.
- Sabò, A.; Doni, M.; Amati, B. SUMOylation of Myc-family proteins. PLoS ONE 2014, 9, e91072.
- Rebello, R.J.; Kusnadi, E.; Cameron, D.P.; Pearson, H.B.; Lesmana, A.; Devlin, J.R.; Drygin, D.; Clark, A.K.; Porter, L.; Pedersen, J.; et al. The dual inhibition of RNA Pol I transcription and PIM kinase as a new therapeutic approach to treat advanced prostate cancer. Clin. Cancer Res. 2016, 22, 5539–5552.
- Höllein, A.; Fallahi, M.; Schoeffmann, S.; Steidle, S.; Schaub, F.X.; Rudelius, M.; Laitinen, I.; Nilsson, L.; Goga, A.; Peschel, C.; et al. Myc-induced SUMOylation is a therapeutic vulnerability for B-cell lymphoma. Blood 2014, 124, 2081–2090.
- Suna, X.X.; Chena, Y.; Sua, Y.; Wanga, X.; Chauhana, K.M.; Lianga, J.; Daniel, C.J.; Sears, R.C.; Dai, M.-S. SUMO protease SENP1 deSUMOylates and stabilizes c-Myc. Proc. Natl. Acad. Sci. USA 2018, 115, 10983–10988.
- Sun, X.X.; Li, Y.; Sears, R.C.; Dai, M.S. Targeting the MYC Ubiquitination-Proteasome Degradation Pathway for Cancer Therapy. Front. Oncol. 2021, 11, 679445.
- Henriksson, M.; Bakardjiev, A.; Klein, G.; Luscher, B. Phosphorylation sites mapping in the N-terminal domain of c-myc modulate its transforming potential. Oncogene 1993, 8, 3199–3209.
- Hann, S.R. Role of post-translational modifications in regulating c-Myc proteolysis, transcriptional activity and biological function. Semin. Cancer Biol. 2006, 16, 288–302.
- Benassi, B.; Fanciulli, M.; Fiorentino, F.; Porrello, A.; Chiorino, G.; Loda, M.; Zupi, G.; Biroccio, A. c-Myc phosphorylation is required for cellular response to oxidative stress. Mol. Cell 2006, 21, 509–519.
- Gregory, M.A.; Qi, Y.; Hann, S.R. Phosphorylation by Glycogen Synthase Kinase-3 Controls c-Myc Proteolysis and Subnuclear Localization. J. Biol. Chem. 2003, 278, 51606–51612.
- Bahram, F.; Von Der Lehr, N.; Cetinkaya, C.; Larsson, L.G. c-Myc hot spot mutations in lymphomas result in inefficient ubiquitination and decreased proteasome-mediated turnover. Blood 2000, 95, 2104–2110.
- Salghetti, S.E.; Kim, S.Y.; Tansey, W.P. Destruction of Myc by ubiquitin-mediated proteolysis: Cancer-associated and transforming mutations stabilize Myc. EMBO J. 1999, 18, 717–726.
- Wang, X.; Cunningham, M.; Zhang, X.; Tokarz, S.; Laraway, B.; Troxell, M.; Sears, R.C. Phosphorylation regulates c-Myc’s oncogenic activity in the mammary gland. Cancer Res. 2011, 71, 925–936.
- Hemann, M.T.; Bric, A.; Teruya-Feldstein, J.; Herbst, A.; Nilsson, J.A.; Cordon-Cardo, C.; Cleveland, J.L.; Tansey, W.P.; Lowe, S.W. Evasion of the p53 tumour surveillance network by tumour-derived MYC mutants. Nature 2005, 436, 807–811.
- Johnson, N.A. Functional and clinical impact of MYC mutations in diffuse large B cell lymphomas. Transl Cancer Res. 2016, 5, S257–S260.
- Huang, Z.; Traugh, J.A.; Bishop, J.M. Negative Control of the Myc Protein by the Stress-Responsive Kinase Pak2. Mol. Cell. Biol. 2004, 24, 1582–1594.
- Macek, P.; Cliff, M.J.; Embrey, K.J.; Holdgate, G.A.; Nissink, J.W.M.; Panova, S.; Waltho, J.P.; Davies, R.A. Myc phosphorylation in its basic helix?loop?helix region destabilizes transient-helical structures, disrupting Max and DNA binding. J. Biol. Chem. 2018, 293, 9301–9310.
- Albihn, A.; Johnsen, J.I.; Henriksson, M.A. MYC in oncogenesis and as a target for cancer therapies. Adv. Cancer Res. 2010, 107, 163–224.
- Herbst, A.; Salghetti, S.E.; Kim, S.Y.; Tansey, W.P. Multiple cell-type-specific elements regulate Myc protein stability. Oncogene 2004, 23, 3863–3871.
- Cowling, V.H.; Chandriani, S.; Whitfield, M.L.; Cole, M.D. A Conserved Myc Protein Domain, MBIV, Regulates DNA Binding, Apoptosis, Transformation, and G 2 Arrest. Mol. Cell. Biol. 2006, 26, 4226–4239.
- Vervoorts, J.; Lüscher-Firzlaff, J.; Lüscher, B. The ins and outs of MYC regulation by posttranslational mechanisms. J. Biol. Chem. 2006, 281, 34725–34729.
- Xiao, D.; Yue, M.; Su, H.; Ren, P.; Jiang, J.; Li, F.; Hu, Y.; Du, H.; Liu, H.; Qing, G. Polo-like Kinase-1 Regulates Myc Stabilization and Activates a Feedforward Circuit Promoting Tumor Cell Survival. Mol. Cell 2016, 64, 493–506.
- Popov, N.; Schülein, C.; Jaenicke, L.A.; Eilers, M. Ubiquitylation of the amino terminus of Myc by SCFβ-TrCP antagonizes SCFFbw7-mediated turnover. Nat. Cell Biol. 2010, 12, 973–981.
- Otto, T.; Horn, S.; Brockmann, M.; Eilers, U.; Schüttrumpf, L.; Popov, N.; Kenney, A.M.; Schulte, J.H.; Beijersbergen, R.; Christiansen, H.; et al. Stabilization of N-Myc Is a Critical Function of Aurora A in Human Neuroblastoma. Cancer Cell 2009, 15, 67–78.
- Jiang, J.; Wang, J.; Yue, M.; Cai, X.; Wang, T.; Wu, C.; Su, H.; Wang, Y.; Han, M.; Zhang, Y.; et al. Direct Phosphorylation and Stabilization of MYC by Aurora B Kinase Promote T-cell Leukemogenesis. Cancer Cell 2020, 37, 200–215.e5.
- Naso, F.D.; Boi, D.; Ascanelli, C.; Pamfil, G.; Lindon, C.; Paiardini, A.; Guarguaglini, G. Nuclear localisation of Aurora-A: Its regulation and significance for Aurora-A functions in cancer. Oncogene 2021, 40, 3917–3928.
- Chan, G.K.L.; Maisel, S.; Hwang, Y.C.; Wolber, R.R.B.; Vu, P.; Patra, C.; Bouhaddou, M.; Kenerson, H.L.; Yeung, R.S.; Swaney, D.L.; et al. Oncogenic PKA signaling stabilizes MYC oncoproteins via an aurora kinase A-dependent mechanism. bioRxiv 2021.
- Zhang, Y.; Wang, Z.; Li, X.; Magnuson, N.S. Pim kinase-dependent inhibition of c-Myc degradation. Oncogene 2008, 27, 4809–4819.
- Zippo, A.; De Robertis, A.; Serafini, R.; Oliviero, S. PIM1-dependent phosphorylation of histone H3 at serine 10 is required for MYC-dependent transcriptional activation and oncogenic transformation. Nat. Cell Biol. 2007, 9, 932–944.
- Devaiah, B.N.; Mu, J.; Akman, B.; Uppal, S.; Weissman, J.D.; Cheng, D.; Baranello, L.; Nie, Z.; Levens, D.; Singer, D.S. MYC protein stability is negatively regulated by BRD4. Proc. Natl. Acad. Sci. USA 2020, 117, 13457–13467.
- Colicino, E.G.; Hehnly, H. Regulating a key mitotic regulator, polo-like kinase 1 (PLK1). Cytoskeleton 2018, 75, 481–494.
- Combes, G.; Alharbi, I.; Braga, L.G.; Elowe, S. Playing polo during mitosis: PLK1 takes the lead. Oncogene 2017, 36, 4819–4827.
- Chiappa, M.; Petrella, S.; Damia, G.; Broggini, M.; Guffanti, F.; Ricci, F. Present and Future Perspective on PLK1 Inhibition in Cancer Treatment. Front. Oncol. 2022, 12, 903016.
- Iliaki, S.; Beyaert, R.; Afonina, I.S. Polo-like kinase 1 (PLK1) signaling in cancer and beyond. Biochem. Pharmacol. 2021, 193, 114747.
- Liu, Z.; Sun, Q.; Wang, X. PLK1, A potential target for cancer therapy. Transl. Oncol. 2017, 10, 22–32.
- Montaudon, E.; Nikitorowicz-Buniak, J.; Sourd, L.; Morisset, L.; EL Botty, R.; Huguet, L.; Dahmani, A.; Painsec, P.; Nemati, F.; Vacher, S.; et al. PLK1 inhibition exhibits strong anti-tumoral activity in CCND1-driven breast cancer metastases with acquired palbociclib resistance. Nat. Commun. 2020, 11, 4053.
- Nieto-Jimenez, C.; Galan-Moya, E.M.; Corrales-Sanchez, V.; Noblejas-Lopez, M.D.M.; Burgos, M.; Domingo, B.; Montero, J.C.; Gomez-Juarez, M.; Picazo-Martinez, M.G.; Esparis-Ogando, A.; et al. Inhibition of the mitotic kinase PLK1 overcomes therapeutic resistance to BET inhibitors in triple negative breast cancer. Cancer Lett. 2020, 491, 50–59.
- Zhang, Z.; Cheng, L.; Li, J.; Qiao, Q.; Karki, A.; Allison, D.B.; Shaker, N.; Li, K.; Utturkar, S.M.; Atallah Lanman, N.M.; et al. Targeting Plk1 sensitizes pancreatic cancer to immune checkpoint therapy. Cancer Res. 2022, 82, 3532–3548.
- Bibi, N.; Parveen, Z.; Rashid, S. Identification of Potential Plk1 Targets in a Cell-Cycle Specific Proteome through Structural Dynamics of Kinase and Polo Box-Mediated Interactions. PLoS ONE 2013, 8, e70843.
- Liu, J.; Zhang, C. The equilibrium of ubiquitination and deubiquitination at PLK1 regulates sister chromatid separation. Cell. Mol. Life Sci. 2017, 74, 2127–2134.
- Raab, M.; Matthess, Y.; Raab, C.A.; Gutfreund, N.; Dötsch, V.; Becker, S.; Sanhaji, M.; Strebhardt, K. A dimerization-dependent mechanism regulates enzymatic activation and nuclear entry of PLK1. Oncogene 2021, 41, 372–386.
- Xu, J.; Shen, C.; Wang, T.; Quan, J. Structural basis for the inhibition of Polo-like kinase 1. Nat Struct Mol Biol. 2013, 20, 1047–1053.
- Beck, J.; Maerki, S.; Posch, M.; Metzger, T.; Persaud, A.; Scheel, H.; Hofmann, K.; Rotin, D.; Pedrioli, P.; Swedlow, J.R.; et al. Ubiquitylation-dependent localization of PLK1 in mitosis. Nat. Cell Biol. 2013, 15, 430–439.
- Kachaner, D.; Garrido, D.; Mehsen, H.; Normandin, K.; Lavoie, H.; Archambault, V. Coupling of Polo kinase activation to nuclear localization by a bifunctional NLS is required during mitotic entry. Nat. Commun. 2017, 8, 1701.
- Zhou, J.; Yang, Q.; Lu, L.; Tuo, Z.; Shou, Z.; Cheng, J. Plk1 inhibition induces immunogenic cell death and enhances immunity against nsclc. Int. J. Med. Sci. 2021, 18, 3516–3525.
- Fu, Z.; Wen, D. The emerging role of polo-like kinase 1 in epithelial-mesenchymal transition and tumor metastasis. Cancers 2017, 9, 131.
- Song, R.; Hou, G.; Yang, J.; Yuan, J.; Wang, C.; Chai, T.; Liu, Z. Effects of PLK1 on proliferation, invasion and metastasis of gastric cancer cells through epithelial-mesenchymal transition. Oncol. Lett. 2018, 16, 5739–5744.
- Gao, Z.; Man, X.; Li, Z.; Bi, J.; Liu, X.; Li, Z.; Li, J.; Zhang, Z.; Kong, C. PLK1 promotes proliferation and suppresses apoptosis of renal cell carcinoma cells by phosphorylating MCM3. Cancer Gene Ther. 2020, 27, 412–423.
- Luo, P.; Yan, H.; Du, J.; Chen, X.; Shao, J.; Zhang, Y.; Xu, Z.; Jin, Y.; Lin, N.; Yang, B.; et al. PLK1 (polo like kinase 1)-dependent autophagy facilitates gefitinib-induced hepatotoxicity by degrading COX6A1 (cytochrome c oxidase subunit 6A1). Autophagy 2021, 17, 3221–3237.
- Oon, M.L.; Hoppe, M.M.; Fan, S.; Phyu, T.; Phuong, H.M.; Tan, S.-Y.; Hue, S.S.-S.; Wang, S.; Poon, L.M.; Chan, H.L.E.; et al. The contribution of MYC and PLK1 expression to proliferative capacity in diffuse large B-cell lymphoma. Leuk Lymphoma 2019, 60, 3214–3224.
- Ren, Y.; Bi, C.; Zhao, X.; Lwin, T.; Wang, C.; Yuan, J.; Silva, A.S.; Shah, B.D.; Fang, B.; Li, T.; et al. PLK1 stabilizes a MYC-dependent kinase network in aggressive B cell lymphomas. J. Clin. Investig. 2018, 128, 5531–5548.
- Yu, Z.; Deng, P.; Chen, Y.; Liu, S.; Chen, J.; Yang, Z.; Chen, J.; Fan, X.; Wang, P.; Cai, Z.; et al. Inhibition of the PLK1-Coupled Cell Cycle Machinery Overcomes Resistance to Oxaliplatin in Colorectal Cancer. Adv. Sci. 2021, 8, 2100759.
- Murga-Zamalloa, C.; Polk, A.; Hanel, W.; Chowdhury, P.; Brown, N.; Hristov, A.C.; Bailey, N.G.; Wang, T.; Phillips, T.; Devata, S.; et al. Polo-like-kinase 1 (PLK-1) and c-myc inhibition with the dual kinase-bromodomain inhibitor volasertib in aggressive lymphomas. Oncotarget 2017, 8, 114474–114480.
- Tan, J.; Li, Z.; Lee, P.L.; Guan, P.; Aau, M.Y.; Lee, S.T.; Feng, M.; Lim, C.Z.; Lee, E.Y.J.; Wee, Z.N.; et al. PDK1 signaling toward PLK1-MYC activation confers oncogenic transformation, tumor-initiating cell activation, and resistance to mTOR-targeted therapy. Cancer Discov. 2013, 3, 1156–1171.
- Padmanabhan, A.; Li, X.; Bieberich, C.J. Protein kinase a regulates MYC protein through transcriptional and post-translational mechanisms in a catalytic subunit isoform-specific manner. J. Biol. Chem. 2013, 288, 14158–14169.
- Wang, D.; Pierce, A.; Veo, B.; Fosmire, S.; Danis, E.; Donson, A.; Venkataraman, S.; Vibhakar, R. A regulatory loop of FBXW7-MYC-PLK1 controls tumorigenesis of MYC-driven medulloblastoma. Cancers 2021, 13, 387.
- Mo, H.; He, J.; Yuan, Z.; Wu, Z.; Liu, B.; Lin, X.; Guan, J. PLK1 contributes to autophagy by regulating MYC stabilization in osteosarcoma cells. Onco Targets Ther. 2019, 12, 7527–7536.
- Quartuccio, S.M.; Schindler, K. Functions of Aurora kinase C in meiosis and cancer. Front. Cell Dev. Biol. 2015, 3, 50.
- Willems, E.; Dedobbeleer, M.; Digregorio, M.; Lombard, A.; Lumapat, P.N.; Rogister, B. The functional diversity of Aurora kinases: A comprehensive review. Cell Div. 2018, 13, 7.
- Joukov, V.; De Nicolo, A. Aurora-PLK1 cascades as key signaling modules in the regulation of mitosis. Sci. Signal. 2018, 11, eaar4195.
- Gallini, S.; Carminati, M.; De Mattia, F.; Pirovano, L.; Martini, E.; Oldani, A.; Asteriti, I.A.; Guarguaglini, G.; Mapelli, M. NuMA phosphorylation by aurora-a orchestrates spindle orientation. Curr. Biol. 2016, 26, 458–469.
- Polverino, F.; Naso, F.D.; Asteriti, I.A.; Palmerini, V.; Singh, D.; Valente, D.; Bird, A.W.; Rosa, A.; Mapelli, M.; Guarguaglini, G. The Aurora-A/TPX2 Axis Directs Spindle Orientation in Adherent Human Cells by Regulating NuMA and Microtubule Stability. Curr. Biol. 2021, 31, 658–667.e5.
- Carmena, M.; Wheelock, M.; Funabiki, H.; Earnshaw, W.C. The chromosomal passenger complex (CPC): From easy rider to the godfather of mitosis. Nat. Rev. Mol. Cell. Biol. 2012, 13, 789–803.
- Van Der Horst, A.; Lens, S.M.A. Cell division: Control of the chromosomal passenger complex in time and space. Chromosoma 2014, 123, 25–42.
- Tang, A.; Gao, K.; Chu, L.; Zhang, R.; Yang, J.; Zheng, J. Aurora kinases: Novel therapy targets in cancers. Oncotarget 2017, 8, 23937–23954.
- Gautschi, O.; Heighway, J.; Mack, P.C.; Purnell, P.R.; Lara, P.N.; Gandara, D.R. Aurora kinases as anticancer drug targets. Clin. Cancer Res. 2008, 14, 1639–1648.
- Lens, S.M.A.; Voest, E.E.; Medema, R.H. Shared and separate functions of polo-like kinases and aurora kinases in cancer. Nat. Rev. Cancer 2010, 10, 825–841.
- Mou, P.K.; Yang, E.J.; Shi, C.; Ren, G.; Tao, S.; Shim, J.S. Aurora kinase A, a synthetic lethal target for precision cancer medicine. Exp. Mol. Med. 2021, 53, 835–847.
- Asteriti, I.A.; Rensen, W.M.; Lindon, C.; Lavia, P.; Guarguaglini, G. The Aurora-A/TPX2 complex: A novel oncogenic holoenzyme? Biochim Biophys Act-Rev. Cancer 2010, 1806, 230–239.
- Wan, X.-B.; Long, Z.-J.; Yan, M.; Xu, J.; Xia, L.-P.; Liu, L.; Zhao, Y.; Huang, X.-F.; Wang, X.-R.; Zhu, X.-F.; et al. Inhibition of Aurora-A suppresses epithelial-mesenchymal transition and invasion by downregulating MAPK in nasopharyngeal carcinoma cells. Carcinogenesis 2008, 29, 1930–1937.
- Liu, X.; Li, Z.; Song, Y.; Wang, R.; Han, L.; Wang, Q.; Jiang, K.; Kang, C.; Zhang, Q. AURKA induces EMT by regulating histone modification through Wnt/ß-catenin and PI3K/Akt signaling pathway in gastric cancer. Oncotarget 2016, 7, 33152–33164.
- Xia, Z.; Wei, P.; Zhang, H.; Ding, Z.; Yang, L.; Huang, Z.; Zhang, N. AURKA governs self-renewal capacity in glioma-initiating cells via stabilization/activation of β-catenin/Wnt Signaling. Mol Cancer Res. 2013, 11, 1101–1111.
- Lin, Z.-Z.; Jeng, Y.-M.; Hu, F.-C.; Pan, H.-W.; Tsao, H.-W.; Lai, P.-L.; Lee, P.-H.; Cheng, A.-L.; Hsu, H.-C. Significance of Aurora B overexpression in hepatocellular carcinoma. Aurora B Overexpression in HCC. BMC Cancer 2010, 10, 461.
- Vischioni, B.; Oudejans, J.J.; Vos, W.; Rodriguez, J.A.; Giaccone, G. Frequent overexpression of aurora B kinase, a novel drug target, in non-small cell lung carcinoma patients. Mol. Cancer Ther. 2006, 5, 2905–2913.
- Qi, G.; Ogawa, I.; Kudo, Y.; Miyauchi, M.; Siriwardena, B.S.M.S.; Shimamoto, F.; Tatsuka, M.; Takata, T. Aurora-B expression and its correlation with cell proliferation and metastasis in oral cancer. Virchows Arch. 2007, 450, 297–302.
- Den Hollander, J.; Rimpi, S.; Doherty, J.R.; Rudelius, M.; Buck, A.; Hoellein, A.; Kremer, M.; Graf, N.; Scheerer, M.; Hall, M.A.; et al. Aurora kinases A and B are up-regulated by Myc and are essential for maintenance of the malignant state. Blood 2010, 116, 1498–1505.
- Dauch, D.; Rudalska, R.; Cossa, G.; Nault, J.C.; Kang, T.-W.; Wuestefeld, T.; Hohmeyer, A.; Imbeaud, S.; Yevsa, T.; Hoenicke, L.; et al. A MYC-aurora kinase A protein complex represents an actionable drug target in p53-altered liver cancer. Nat. Med. 2016, 22, 744–753.
- Beltran, H.; Rickman, D.S.; Park, K.; Chae, S.S.; Sboner, A.; MacDonald, T.Y.; Wang, Y.; Sheikh, K.L.; Terry, S.; Tagawa, S.T.; et al. Molecular characterization of neuroendocrine prostate cancer and identification of new drug targets. Cancer Discov. 2011, 1, 487–495.
- Vader, G.; Lens, S.M.A. The Aurora kinase family in cell division and cancer. Biochim. Biophys. Acta-Rev. Cancer 2008, 1786, 60–72.
- Büchel, G.; Carstensen, A.; Mak, K.-Y.; Roeschert, I.; Leen, E.; Sumara, O.; Hofstetter, J.; Herold, S.; Kalb, J.; Baluapuri, A.; et al. Association with Aurora-A Controls N-MYC-Dependent Promoter Escape and Pause Release of RNA Polymerase II during the Cell Cycle. Cell Rep. 2017, 21, 3483–3497.
- Richards, M.W.; Burgess, S.G.; Poon, E.; Carstensen, A.; Eilers, M.; Chesler, L.; Bayliss, R. Structural basis of N-Myc binding by Aurora-A and its destabilization by kinase inhibitors. Proc. Natl. Acad. Sci. USA 2016, 113, 13726–13731.
- Lu, L.; Han, H.; Tian, Y.; Li, W.; Zhang, J.; Feng, M.; Li, Y. Aurora kinase A mediates c-Myc’s oncogenic effects in hepatocellular carcinoma. Mol. Carcinog. 2015, 54, 1467–1479.
- Zheng, F.; Yue, C.; Li, G.; He, B.; Cheng, W.; Wang, X.; Yan, M.; Long, Z.; Qiu, W.; Yuan, Z.; et al. Nuclear AURKA acquires kinase-independent transactivating function to enhance breast cancer stem cell phenotype. Nat. Commun. 2016, 7, 10180.
- Hsueh, K.W.; Fu, S.L.; Huang, C.Y.F.; Lin, C.H. Aurora-A phosphorylates hnRNPK and disrupts its interaction with p53. FEBS Lett. 2011, 585, 2671–2675.
- Ho, J.S.L.; Ma, W.; Mao, D.Y.L.; Benchimol, S. p53-Dependent Transcriptional Repression of c-myc Is Required for G1 Cell Cycle Arrest. Mol. Cell. Biol. 2005, 25, 7423–7431.
- Santoro, A.; Vlachou, T.; Luzi, L.; Melloni, G.; Mazzarella, L.; D’Elia, E.; Aobuli, X.; Pasi, C.E.; Reavie, L.; Bonetti, P.; et al. p53 Loss in Breast Cancer Leads to Myc Activation, Increased Cell Plasticity, and Expression of a Mitotic Signature with Prognostic Value. Cell Rep. 2019, 26, 624–638.e8.
- Bayliss, R.; Sardon, T.; Ebert, J.; Lindner, D.; Vernos, I.; Conti, E. Determinants for Aurora-A activation and Aurora-B discrimination by TPX2. Cell Cycle 2004, 3, 402–405.
- Fu, J.; Bian, M.; Liu, J.; Jiang, Q.; Zhang, C. A single amino acid change converts Aurora-A into Aurora-B-like kinase in terms of partner specificity and cellular function. Proc. Natl. Acad. Sci. USA 2009, 106, 6939–6944.
- Eyers, P.A.; Churchill, M.E.A.; Maller, J.L. The Aurora A and Aurora B protein kinases: A single amino acid difference controls intrinsic activity and activation by TPX2. Cell Cycle 2005, 4, 784–789.
- Hans, F.; Skoufias, D.A.; Dimitrov, S.; Margolis, R.L. Molecular distinctions between Aurora A and B: A single residue change transforms Aurora A into correctly localized and functional Aurora B. Mol. Biol. Cell 2009, 20, 3491–3502.
- DeLuca, K.F.; Meppelink, A.; Broad, A.J.; Mick, J.E.; Peersen, O.B.; Pektas, S.; Lens, S.M.; DeLuca, J.G. Aurora A kinase phosphorylates Hec1 to regulate metaphase kinetochore-microtubule dynamics. J. Cell Biol. 2018, 217, 163–177.
- Berwanger, B.; Hartmann, O.; Bergmann, E.; Bernard, S.; Nielsen, D.; Krause, M.; Kartal, A.; Flynn, D.; Wiedemeyer, R.; Schwab, M.; et al. Loss of a FYN-regulated differentiation and growth arrest pathway in advanced stage neuroblastoma. Cancer Cell 2002, 2, 377–386.
- Brockmann, M.; Poon, E.; Berry, T.; Carstensen, A.; Deubzer, H.E.; Rycak, L.; Jamin, Y.; Thway, K.; Robinson, S.P.; Roels, F.; et al. Small Molecule Inhibitors of Aurora-A Induce Proteasomal Degradation of N-Myc in Childhood Neuroblastoma. Cancer Cell 2013, 24, 75–89.
- Bogen, D.; Wei, J.S.; Azorsa, D.O.; Ormanoglu, P.; Buehler, E.; Guha, R.; Keller, J.M.; Griner, L.A.M.; Ferrer, M.; Song, Y.K.; et al. Aurora B kinase is a potent and selective target in MYCN-driven neuroblastoma. Oncotarget 2015, 6, 35247–35262.
- Borah, N.A.; Sradhanjali, S.; Barik, M.R.; Jha, A.; Tripathy, D.; Kaliki, S.; Rath, S.; Raghav, S.K.; Patnaik, S.; Mittal, R.; et al. Aurora kinase B expression, its regulation and therapeutic targeting in human retinoblastoma. Investig. Ophthalmol. Vis. Sci. 2021, 62, 16.
- Li, M.; Sun, C.; Bu, X.; Que, Y.; Zhang, L.; Zhang, Y.; Zhang, L.; Lu, S.; Huang, J.; Zhu, J.; et al. ISL1 promoted tumorigenesis and EMT via Aurora kinase A-induced activation of PI3K/AKT signaling pathway in neuroblastoma. Cell Death Dis. 2021, 12, 620.
- Dar, A.A.; Belkhiri, A.; El-Rifai, W. The aurora kinase A regulates GSK-3β in gastric cancer cells. Oncogene 2009, 28, 866–875.
- Katayama, H.; Sasai, K.; Kawai, H.; Yuan, Z.-M.; Bondaruk, J.; Suzuki, F.; Fujii, S.; Arlinghaus, R.B.; Czerniak, B.A.; Sen, S. Phosphorylation by aurora kinase A induces Mdm2-mediated destabilization and inhibition of p53. Nat. Genet. 2004, 36, 55–62.
More