Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Sirius Huang and Version 1 by Jie-Li Hu.
Eradication of covalently closed circular DNA (cccDNA) is an ideal goal of chronic hepatitis B (CHB) therapy. Understanding the changes in the cccDNA pool during therapy provides a basis for developing CHB treatment strategies. Central to the description of cccDNA dynamics is a parameter called cccDNA half-life. CccDNA half-life is not an intrinsic property of cccDNA molecules, but a description of an observed phenomenon characterized by cccDNA pool decline. Since cccDNA has to be in the nuclei of host cells to function, the half-life of cccDNA is determined by the state and destiny of the host cells.
- hepatitis B virus
- cccDNA
- half-life
- nucleot(s)ide analogues
- hepatocyte
- turnover
1. Introduction
Covalently closed circular DNA (cccDNA) is the first viral product after hepatitis B virus (HBV) infection of hepatocytes. It serves as a stable repository of HBV genetic information in liver cells and represents the most impenetrable barrier to a functional cure for chronic hepatitis B (CHB) [1,2,3][1][2][3]. CccDNA is converted from its precursor, relaxed circular DNA (rcDNA) (Figure 1). With great effort, in almost two decades, researchers have largely characterized the host factors required for cccDNA formation, intermediates formed during the conversion and the functional regulation mechanisms of cccDNA minichromosomes (see reviews [4,5,6][4][5][6]). There is a consensus that the functional cure of CHB requires eradication or persistent suppression of cccDNA [2,7,8,9,10][2][7][8][9][10]. However, current therapies based on the nucleot(s)ide analog (NUC) or/and pegylated-interferon-α (PEG-IFNα) have limited efficacy in achieving a functional cure for CHB, necessitating the development of new therapeutic strategies [11,12,13,14,15,16][11][12][13][14][15][16].

Figure 1. Replication cycle of HBV. The viruses infect hepatocytes by interacting with the receptor sodium taurocholate cotransporting polypeptide (NTCP). Relaxed circular DNA (rcDNA) is released into the nuclei after nucleocapsid uncoating. With the help of the host factors, rcDNA is converted into cccDNA, which serves as the template of viral RNA transcription. The productive nucleocapsid containing progeny rcDNA either secretes through the multivesicular body (MVB) pathway or recycles to the nuclei.
It is well known that serum HBV virions are in a highly dynamic equilibrium; approximately 1011 viruses decay and are produced daily [17]. Similarly, the cccDNA pool in hepatocytes is in a dynamic equilibrium [18]. Since any treatment to cure CHB must reduce the number of cccDNA molecules in the livers, it is important to understand the dynamic changes in cccDNA quantity during therapy. In fact, cccDNA dynamics can only be revealed by treatments that shift the equilibrium in the cccDNA pool. Both NUC and PEG-IFNα can reduce cccDNA levels either indirectly by suppressing cccDNA synthesis or directly by degrading cccDNA. The decline of cccDNA during therapy denotes a shift in the dynamic balance between cccDNA synthesis and decay. The dynamics of the cccDNA pool can be investigated by exploring this shift.
2. The Term ‘cccDNA Half-Life’ Does Not Refer to an Intrinsic Property of cccDNA
The half-life of pure cccDNA independent of its host cells is practically meaningless, since cccDNA has to be in the nuclei of host cells in order to function. It is the destiny and environment of the host cells that govern the ‘half-life’ of cccDNA. The cccDNA disappear when the host cells die, and decrease when the host cells divide. CccDNA can also be degraded in noncytopathic ways. The cccDNA pool may exhibit very different half-lives under different conditions. In chimpanzees recovering from an acute HBV infection, cccDNA can decline with a half-life as short as 3 days [19]. In contrast, patients receiving long-term NUC therapy exhibited a very slow cccDNA decline, with a half-life of as long as 26 months [20]. Apparently, ‘cccDNA half-life’ involves the quantified amplitudes of the cccDNA decline phenomenon, which vary considerably depending on the situation. Therefore, this term does not refer to the intrinsic nature of cccDNA, and that the values represent the observed kinetics of cccDNA decay as a pool that would change depending on the circumstances. In this sense, the term ‘apparent cccDNA half-life’, as proposed by Boettler et al., is more accurate than ‘cccDNA half-life’ [18].
3. Major Factors That Affect cccDNA Half-Life
Given that cccDNA is located in the nuclei of the host cells and performs its functions, the state of the host cells will exert a significant influence on the dynamics of cccDNA. Therefore, factors affecting the situations of host cells affect cccDNA half-life. Noncytopathic effects and hepatocyte turnover are the major factors impacting cccDNA half-life.
3.1. Noncytopathic Effects Contribute to cccDNA Degradation in Acute Infection
Chisari et al. first reported that HBV-specific cytotoxic T lymphocytes could abolish HBV gene expression and replication in the liver of transgenic mice via noncytopathic cytokines [21]. Based on these findings, they postulated that this antiviral process might be primarily responsible for the viral clearance during human HBV infection rather than the destruction of infected cells. To test this hypothesis, they observed the clearance of HBV in acutely infected chimpanzees. In these animals, a significant decrease in HBV replicative intermediates and cccDNA from the liver was observed long before the peak of T cell infiltration and most of the liver disease. Xia et al. also found that HBV-specific T cells inhibit HBV replication and reduce cccDNA in infected cells without the direct contact required for cytolysis [22]. The findings support the presence of a noncytopathic mechanism contributing to the clearance of the virus [23]. Mathematical modeling was later performed to evaluate the extent to which cytopathic and noncytopathic T cell effector functions contribute to the resolution of HBV infection in three acutely infected chimpanzees. CccDNA demonstrated a rapid decay in the first phase of the decline, with a half-life of 3 days. If cell death and cell division were the only mechanisms responsible for clearance of HBV infection, such a short half-life would imply the destruction and regeneration of approximately 11 livers. In contrast, simulation incorporating the cytokine effects indicated significantly less hepatocyte death and regeneration (1.4–2.8 livers) [19]. Studies using woodchuck models also supported that both noncytopathic effects and hepatocyte death were responsible for the elimination of cccDNA during recovery from transient infections [24]. Cytokines associated with this noncytopathic effect include interferon-gamma (IFNγ), tumor necrosis factor-alpha (TNFα), and IFNα/β [22,25][22][25]. Later studies showed that IFNα accelerates the decay of DHBV cccDNA in culture [26] and induces the specific degradation of the nuclear HBV cccDNA without hepatotoxicity by up-regulating APOBEC3A and APOBEC3B cytidine deaminases [22,27][22][27].
3.2. Hepatocyte Turnover Is a Major Factor Promoting cccDNA Decay
The liver is a solid organ with a high regenerative capacity to ensure that the liver-to-bodyweight ratio is always at 100% of what is required for body homeostasis [28]. Hepatocyte turnover is regulated by two closely related events, namely hepatocyte death and division. As hepatocytes die, the number of hepatocytes must be replenished to maintain liver mass and new hepatocytes are produced. Typically, a cell death results in a new cell being produced, and vice versa. It is believed that cccDNA is degraded or eliminated when the host cell dies and cannot infect another hepatocyte.
3.2.1. Evidence That Hepatocyte Turnover Promotes cccDNA Decay
The influence of hepatocyte turnover on cccDNA levels (or infected hepatocytes) was first demonstrated by Mason et al. [29]. An NUC, 2′-deoxycarbocyclic guanosine (2′-CDG), was found to significantly reduce DHBV cccDNA levels (with a half-life of approximately 2 weeks) in the livers of ducks congenitally infected with DHBV. 5-bromo-2’-deoxyuridine (BrdU) labelling showed that hepatocytes proliferation increased by 10-fold after 2 weeks of therapy. This result suggests that the inhibition of viral replication and acceleration of hepatocyte turnover with 2′-CDG therapy caused a rapid clearance of infected hepatocytes. In contrast, 5-fluoro-2’,3’-dideoxy-3’-thiacytidine (524 W), another potent inhibitor of DHBV DNA synthesis that did not expedite the turnover of hepatocytes in ducks, led to a strong inhibition of virus production but a slower rate of decline in the number of infected hepatocytes and cccDNA (with a half-life of more than 6 weeks) than that with 2′-CDG. Addison et al. analyzed the decline of DHBV cccDNA in ducks treated with a combination of lamivudine (LAM) and a dideoxyguanosine prodrug for 5 months [30]. The cccDNA pools in three ducks demonstrated an exponential decline, with half-lives ranging from 35 to 57 days. Liver sections stained with the cell division marker, proliferating cell nuclear antigen (PCNA), showed that animals with a continuous cccDNA loss had a significantly higher number of PCNA-positive nuclei than those whose cccDNA levels had plateaued.
Mason et al. also treated WHV-infected primary hepatocyte cultures with [1-(2-fluoro-5-methyl-β-L-arabinofuranosyl) uracil] (L-FMAU), a reverse transcriptase inhibitor. They found that although L-FMAU caused a 200-fold suppression in viral DNA replication, no significant loss in cccDNA was observed in the infected hepatocytes during the 40-day treatment [31]. In chronically infected woodchucks, L-FMAU treatment for 30 weeks reduced cccDNA levels to between 1.2 and 5.4% of pretreatment levels, suggesting a half-life of 33 to 50 days. This reduction could be explained by an infected-cell death rate of 1.3 to 2.1% per day, which is in reasonable agreement with the PCNA staining results.
Outstanding examples exhibiting the influence of hepatocyte death on cccDNA is from the experimental therapy of HBV infection by T cells that were engineered to express HBV-specific chimeric antigen receptors (CARs) or T cell receptors (TCRs). Protzer’s lab reported that expression of CARs directed against the HBV surface proteins enables human T cells to kill HBV-infected human hepatocytes and to eliminate viral cccDNA in vitro and in vivo [32,33][32][33]. T cells stably expressing high-affinity HBV envelope- or core-specific TCRs also effectively control HBV infection in humanized mice by specifically clearing infected hepatocytes without damaging noninfected cells [34].
3.2.2. Effect of Cell Division on cccDNA
In nonproliferating primary cultures of woodchuck hepatocyte infected with woodchuck hepatitis virus (WHV), treatment of L-FMAU did not facilitate loss of cccDNA. No significant loss of cccDNA from the treated cultures was seen between days 8 and 40 post-infection, indicating a cccDNA half-life of at least 32 days [31]. Ko et al. tested cccDNA half-life in an HBV-infected cell line (HepG2-NTCP-K7) in the presence of 2.5% DMSO, which induces cell cycle arrest. Treating cells with entecavir (ETV) reduced cccDNA levels by 48% at day 45 post-infection, suggesting a cccDNA half-life of approximately 40 days in these nondividing cells [35].
During hepatocyte division, the HBV cccDNA minichromosome could be distributed unequally or even lost during mitosis since it is not a cellular chromosome equipped with centromere structures. Chong et al. examined the dynamic changes in HBV cccDNA in different cellular growth stages of a stably HBV-producing cell line, 1.3ES2 [36]. They found that the amount of cccDNA decreased dramatically in the cells during their exponential proliferation, and cccDNA could be removed when proliferating cells were subjected to long term of lamivudine (3TC) treatment. The half-life of cccDNA in the exponentially proliferating cells was approximately 5 days. Once the cells had grown to confluence, the half-life of cccDNA was approximately 9 days, but 20% of the remaining cccDNA was retained stably inside the cells for more than 30 days [36]. To quantify the impact of cell division on cccDNA loss, Tu et al. passaged cells every 3 days to induce mitosis of HBV-infected HepG2-NTCP and HepaRG-NTCP cells. Then, the number of cccDNA copies was measured by precise PCR assays and the number of HBV-expressing cells was monitored over time using reporter viruses. They observed that cccDNA levels undergo a 5-fold decrease after each round of mitosis, which is the exact rate predicted by mathematical models assuming a complete loss of cccDNA in daughter cells [37].
In contrast, some other studies did not observe significant cccDNA loss during cell division. Li et al. determined the distribution of HBV cccDNA in continuously passaged HepAD38 cells using a fluorescence imaging in situ hybridization (FISH)-based assay. The findings showed that nuclear HBV DNA symmetrically distribute to daughter cells. It is worth noting that the detected nuclear DNA was not necessarily cccDNA, since the probes used were not cccDNA-specific [38]. Dandri et al. evaluate whether nuclear cccDNA becomes unstable during cell division in cultures of primary hepatocytes isolated from a WHV chronically infected woodchuck [39]. The cells were treated with epidermal growth factor (EGF) for 24 days to induce proliferation in the presence of adefovir (ADV). They found that cccDNA signals had the same intensity in ADV-treated plates despite the fact that a 50% increase in cell number (70% vs. 105%) did occur when EGF was added. Cautions should be taken while interpreting this result, because the cells seemed to proliferate at a marginal rate during a long period.
To determine the effect of cell proliferation on cccDNA in vivo, Lutgehetmann et al. transplanted primary tupaia hepatocytes (PTHs) chronically infected with woolly monkey HBV (WM-HBV) from chimeric mice into the urokinase-type plasminogen activator (uPA)/severe combined immunodeficiency (SCID)/beige (USB) mouse model. Transplantation of WM-HBV-infected hepatocytes led to an average of 3.8 PTH doublings within 80 days. Remarkably, a median 2-log decline of cccDNA per cell determined during PTH proliferation was due to both dilution of the cccDNA pool among daughter cells and a 0.5-log loss of intrahepatic cccDNA loads [40]. The effect of hepatocyte division on the HBV cccDNA pool was also observed in humanized mice. Primary human hepatocytes (PHH) from HBV-infected humanized mice were serially transplanted into naïve recipients. The findings demonstrated that human hepatocyte division, triggers substantial cccDNA loss even without the involvement of cytolytic mechanisms. CccDNA copies per PHH decreased by 2.4 log during cell division (from day 3 post-transplantation until day 30), and the total cccDNA amounts per liver reduced by 0.7 log during this period [41]. In contrast, Hayashi et al. demonstrated that in HBV-infected human-liver-chimeric mice (PXB-mice), the total cccDNA content did not change during liver repopulation after entecavir treatment. An explanation for the inconsistence is that the experimental procedure used by Hayashi et al. is different from that used by Allweiss et al. In PXB-mice, observation started 8 weeks after transplantation and 4 weeks after HBV infection. Allweiss et al. adopted a different procedure in which human liver cells from USB mice already infected with HBV were transplanted to naïve recipients and observation started 3 days after transplantation. Probably, this procedure maximized the proliferation rate of the cells and thus presented its effect on cccDNA more readily.
3.2.3. Hepatocyte Turnover Rate
Knowing the lifespan or turnover rate of hepatocytes is essential in exploring the dynamics of the cccDNA pool due to the close relationship between cell death and the change in the cccDNA pool. In 1961, Richard determined the lifespan of rat liver cells by tracing tritiated thymidine (H3-thymidine) incorporated into cell nuclei [42]. He found that 0.22% of hepatic nuclei were labelled following a single injection of H3-thy-midine in normal adult rats, suggesting that a liver would renew in 450 days. Of note, it was estimated that in normal liver approximately 41% of labeled cells had died or divided by 60 days, and the remaining labeled cells had lifespans varying up to at least 6 additional months [42] (Figure 2A). This implies a great heterogeneity of hepatocytes lifespan. For fatty and cirrhotic liver, the lifespan of hepatocytes was considerably less than that of normal liver (26 days vs. 450 days). Again, those hepatocytes exhibited heterogeneity in lifespan. Although 98% of labelled hepatocytes died or divided in 60 days, the remaining 2% of cells did not go further change for at least several months. Magami et al. used H3-thymidine to trace the life of hepatocytes in mice [43]. They found that the proportion of labelled hepatocytes decreased from 33.7% to 16.6%, 10.9% and 7.9% after 100, 200 and 300 days (Figure 2A). This indicates an overall hepatocyte half-life of approximately 100 days, but the heterogeneity is apparent at individual cell level, given that 7.9% of the cells survived longer than 300 days [43].
The concept of the balance between hepatocyte death and production provides a basis for estimating hepatocyte death rate by monitoring newly produced hepatocytes over time. Recently, He et al. assessed hepatocyte proliferation in a mouse model using Ki67-induced expression of the fluorescent protein [44]. Their dual recombinase-based method, called Protracer (proliferation tracer, Figure 2B), enabled the continuous recording of the proliferation events of entire cell populations over time in multiple organs, including the liver. Although their primary goal was to characterize the cell source of hepatocytes during homeostasis and regeneration, the study provided valuable information to estimate the turnover of hepatocytes. One set of data revealed that from week 2 to week 12 after tamoxifen induction, a gradual increase in the number of newly produced hepatocytes was observed, as demonstrated by the GFP expression in the cells induced by Ki67 expression. At week 12, over 60% of the total population were GFP-positive cells (Figure 2C). This implies that more than 60% of the hepatocytes died during the course of the 10 weeks, as approximately 60% of the hepatocytes were newly produced at this time. Given that some GFP-positive hepatocytes must have already died, 10 weeks may be a low estimate of the half-life of mouse hepatocytes in homeostasis. This estimate closes to the half-life of hepatocytes (100 days) inferred from the H3-thymidine labelling experiments. In addition, different regions of the liver lobule contribute differentially to hepatocyte turnover, and zone 2 is the primary source of new hepatocytes during homeostasis and regeneration (Figure 2D) [44,45][44][45].
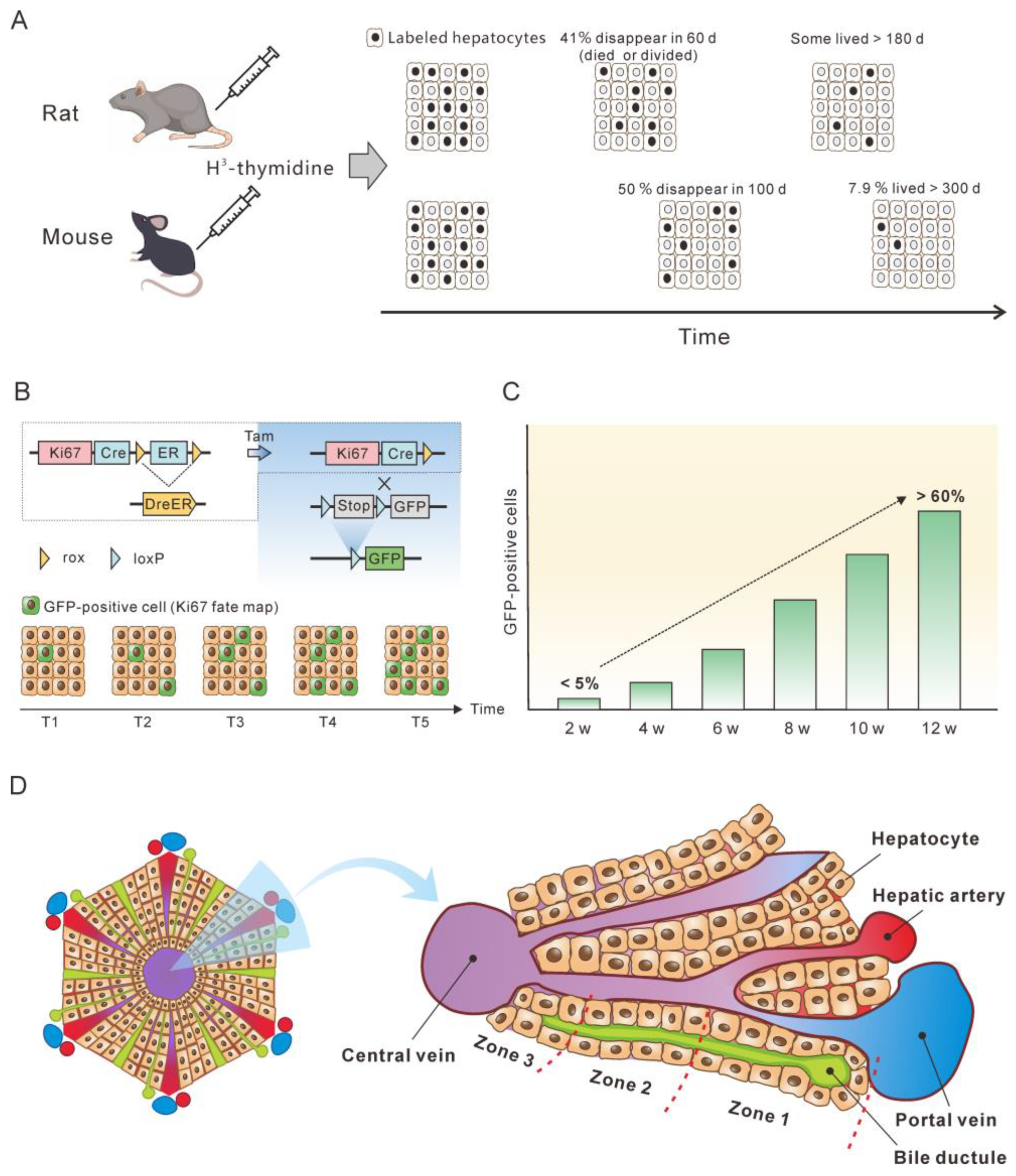
Figure 2. Monitoring the lifespan and proliferation of hepatocytes. (A) H3-thymidine was injected into rat and mouse models to label hepatocytes and monitor the changes in the labeled cells [42,43][42][43]. (B) Ki67 expression induced a constitutive expression of GFP which was used to monitor the fate of liver cells. (C) From week 2 to week 12, the number of hepatocytes with Ki67-expression-induced GFP expression increased to more than 50%, which represent newly produced hepatocytes. The figure is a modification of a figure from reference [45]. (D) Structure of the liver lobule. Hepatocytes in the liver lobule are organized into three zones and those in zones 2 and 1 have higher proliferation rates than those in zone 3 during homeostasis.
The half-life of normal human hepatocytes does not permit experimental determination in vivo by lineage or H3-thymidine tracing. However, indirect estimation of normal human hepatocyte turnover can be made by immunohistochemical detection of PCNA. PCNA is present in the cell nuclei throughout the cell cycle but binds tightly to chromatin at the peak of the S-phase. Immunofluorescent studies have shown that in cells only PCNA associated with DNA replication sites (S-phase-specific PCNA) can be detected. In contrast, non-S-phase nuclear PCNA, which is present in lower amounts and is not physically associated with DNA replication sites, is likely lost or undetectable by conventional immunocytochemical methods [46,47,48][46][47][48]. In methanol-fixed normal human liver, the PCNA-labeling index was from 0.05% to 0.78% for hepatocytes [49,50][49][50]. This translates to 0.15 to 2.34% of hepatocyte death per day, assuming that S-phase lasts for approximately 8 h and that PCNA staining therefore only reflects approximately one-third of the cells in S-phase over a 24 h period [51]. Another proliferation marker, Ki67, can also be used to estimate hepatocyte turnover rate. Farinati et al. found that 0.2% of the hepatocytes were Ki6-positive in the periportal area (zone 1) in the samples from patients with hepatitis B, translating to a 0.6% of hepatocyte death per day. However, this might be a low estimate since the majority of proliferating hepatocytes were found in zone 2 of mouse lobules [44,45][44][45].
In summary, normal hepatocytes of rat and mice exhibit a heterogeneity in half-life. The majority of hepatocytes have a half-life of 50–100 days, but a minority of hepatocytes can live longer than 300 days. The death rate of normal human hepatocytes is estimated to be approximately 0.5% per day, equivalent to a half-life of 100 days. However, details of half-life heterogeneity of human hepatocytes need further investigation.
References
- Lok, A.S.; Zoulim, F.; Dusheiko, G.; Ghany, M.G. Hepatitis B cure: From discovery to regulatory approval. Hepatology 2017, 66, 1296–1313.
- Ning, Q.; Wu, D.; Wang, G.-Q.; Ren, H.; Gao, Z.-L.; Hu, P.; Han, M.-F.; Wang, Y.; Zhang, W.-H.; Lu, F.-M.; et al. Roadmap to functional cure of chronic hepatitis B: An expert consensus. J. Viral Hepat. 2019, 26, 1146–1155.
- Wong, G.L.H.; Gane, E.; Lok, A.S.F. How to achieve functional cure of HBV: Stopping NUCs, adding interferon or new drug de-velopment? J. Hepatol. 2022, 76, 1249–1262.
- Wei, L.; Ploss, A. Mechanism of Hepatitis B Virus cccDNA Formation. Viruses 2021, 13, 1463.
- Xia, Y.; Guo, H. Hepatitis B virus cccDNA: Formation, regulation and therapeutic potential. Antivir. Res. 2020, 180, 104824.
- Hong, X.; Kim, E.S.; Guo, H. Epigenetic regulation of hepatitis B virus covalently closed circular DNA: Implications for epigenetic therapy against chronic hepatitis B. Hepatology 2017, 66, 2066–2077.
- Block, T.M.; Alter, H.; Brown, N.; Brownstein, A.; Brosgart, C.; Chang, K.-M.; Chen, P.-J.; Cohen, C.; El-Serag, H.; Feld, J.; et al. Research priorities for the discovery of a cure for chronic hepatitis B: Report of a workshop. Antivir. Res. 2018, 150, 93–100.
- Martinez, M.G.; Boyd, A.; Combe, E.; Testoni, B.; Zoulim, F. Covalently closed circular DNA: The ultimate therapeutic target for curing HBV infections. J. Hepatol. 2021, 75, 706–717.
- Lim, S.G.; Baumert, T.F.; Boni, C.; Gane, E.; Levrero, M.; Lok, A.S.; Maini, M.K.; Terrault, N.A.; Zoulim, F. The scientific basis of combination therapy for chronic hepatitis B functional cure. Nat. Rev. Gastroenterol. Hepatol. 2023, 1–16.
- Dusheiko, G.; Agarwal, K.; Maini, M.K. New Approaches to Chronic Hepatitis B. N. Engl. J. Med. 2023, 388, 55–69.
- Degasperi, E.; Anolli, M.P.; Lampertico, P. Towards a Functional Cure for Hepatitis B Virus: A 2022 Update on New Antiviral Strategies. Viruses 2022, 14, 2404.
- Zoulim, F. Inhibition of hepatitis B virus gene expression: A step towards functional cure. J. Hepatol. 2018, 68, 386–388.
- Mak, L.-Y.; Cheung, K.-S.; Fung, J.; Seto, W.-K.; Yuen, M.-F. New strategies for the treatment of chronic hepatitis B. Trends Mol. Med. 2022, 28, 742–757.
- Fanning, G.C.; Zoulim, F.; Hou, J.; Bertoletti, A. Therapeutic strategies for hepatitis B virus infection: Towards a cure. Nat. Rev. Drug Discov. 2019, 18, 827–844.
- Naggie, S.; Lok, A.S. New Therapeutics for Hepatitis B: The Road to Cure. Annu. Rev. Med. 2021, 72, 93–105.
- Ma, Z.; Zhang, E.; Gao, S.; Xiong, Y.; Lu, M. Toward a Functional Cure for Hepatitis B: The Rationale and Challenges for Therapeutic Targeting of the B Cell Immune Response. Front. Immunol. 2019, 10, 2308.
- Nowak, M.A.; Bonhoeffer, S.; Hill, A.M.; Boehme, R.; Thomas, H.C.; McDade, H. Viral dynamics in hepatitis B virus infection. Proc. Natl. Acad. Sci. USA 1996, 93, 4398–4402.
- Boettler, T.; Gill, U.S.; Allweiss, L.; Pollicino, T.; Tavis, J.E.; Zoulim, F. Assessing immunological and virological responses in the liver: Implications for the cure of chronic hepatitis B virus infection. JHEP Rep. 2022, 4, 100480.
- Murray, J.M.; Wieland, S.F.; Purcell, R.H.; Chisari, F.V. Dynamics of hepatitis B virus clearance in chimpanzees. Proc. Natl. Acad. Sci. USA 2005, 102, 17780–17785.
- Boyd, A.; Lacombe, K.; Lavocat, F.; Maylin, S.; Miailhes, P.; Lascoux-Combe, C.; Delaugerre, C.; Girard, P.M.; Zoulim, F. Decay of ccc-DNA marks per-sistence of intrahepatic viral DNA synthesis under tenofovir in HIV-HBV co-infected patients. J. Hepatol. 2016, 65, 683–691.
- Guidotti, L.G.; Ishikawa, T.; Hobbs, M.V.; Matzke, B.; Schreiber, R.; Chisari, F.V. Intracellular Inactivation of the Hepatitis B Virus by Cytotoxic T Lymphocytes. Immunity 1996, 4, 25–36.
- Xia, Y.; Stadler, D.; Lucifora, J.; Reisinger, F.; Webb, D.; Hösel, M.; Michler, T.; Wisskirchen, K.; Cheng, X.; Zhang, K.; et al. Interferon-gamma and Tumor Necrosis Factor-alpha Produced by T Cells Reduce the HBV Persistence Form, cccDNA, Without Cytolysis. Gastroenterology 2016, 150, 194–205.
- Guidotti, L.G.; Rochford, R.; Chung, J.; Shapiro, M.; Purcell, R.; Chisari, F.V. Viral Clearance Without Destruction of Infected Cells During Acute HBV Infection. Science 1999, 284, 825–829.
- Mason, W.S.; Xu, C.; Low, H.C.; Saputelli, J.; Aldrich, C.E.; Scougall, C.; Grosse, A.; Colonno, R.; Litwin, S.; Jilbert, A.R. The amount of hepatocyte turnover that oc-curred during resolution of transient hepadnavirus infections was lower when virus replication was inhibited with entecavir. J. Virol. 2009, 83, 1778–1789.
- Guidotti, L.G.; Ando, K.; Hobbs, M.V.; Ishikawa, T.; Runkel, L.; Schreiber, R.D.; Chisari, F.V. Cytotoxic T lymphocytes inhibit hepatitis B virus gene expression by a noncytolytic mechanism in transgenic mice. Proc. Natl. Acad. Sci. USA 1994, 91, 3764–3768.
- Liu, F.; Campagna, M.; Qi, Y.; Zhao, X.; Guo, F.; Xu, C.; Li, S.; Li, W.; Block, T.M.; Chang, J.; et al. Alpha-Interferon Suppresses Hepadnavirus Transcription by Altering Epigenetic Modification of cccDNA Minichromosomes. PLoS Pathog. 2013, 9, e1003613.
- Lucifora, J.; Xia, Y.; Reisinger, F.; Zhang, K.; Stadler, D.; Cheng, X.; Sprinzl, M.F.; Koppensteiner, H.; Makowska, Z.; Volz, T.; et al. Specific and Nonhepatotoxic Degradation of Nuclear Hepatitis B Virus cccDNA. Science 2014, 343, 1221–1228.
- Michalopoulos, G.K.; Bhushan, B. Liver regeneration: Biological and pathological mechanisms and implications. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 40–55.
- Fourel, I.; Cullen, J.M.; Saputelli, J.; Aldrich, C.E.; Schaffer, P.; Averett, D.R.; Pugh, J.; Mason, W.S. Evidence that hepatocyte turnover is required for rapid clearance of duck hepatitis B virus during antiviral therapy of chronically infected ducks. J. Virol. 1994, 68, 8321–8330.
- Addison, W.R.; Walters, K.A.; Wong, W.W.; Wilson, J.S.; Madej, D.; Jewell, L.D.; Tyrrell, D.L. Half-life of the duck hepatitis B virus co-valently closed circular DNA pool in vivo following inhibition of viral replication. J. Virol. 2002, 76, 6356–6363.
- Zhu, Y.; Yamamoto, T.; Cullen, J.; Saputelli, J.; Aldrich, C.E.; Miller, D.S.; Litwin, S.; Furman, P.A.; Jilbert, A.; Mason, W.S. Kinetics of Hepadnavirus Loss from the Liver during Inhibition of Viral DNA Synthesis. J. Virol. 2001, 75, 311–322.
- Bohne, F.; Chmielewski, M.; Ebert, G.; Wiegmann, K.; Kürschner, T.; Schulze, A.; Urban, S.; Krönke, M.; Abken, H.; Protzer, U. T Cells Redirected Against Hepatitis B Virus Surface Proteins Eliminate Infected Hepatocytes. Gastroenterology 2008, 134, 239–247.
- Krebs, K.; Böttinger, N.; Huang, L.; Chmielewski, M.; Arzberger, S.; Gasteiger, G.; Jäger, C.; Schmitt, E.; Bohne, F.; Aichler, M.; et al. T Cells Expressing a Chimeric Antigen Receptor That Binds Hepatitis B Virus Envelope Proteins Control Virus Replication in Mice. Gastroenterology 2013, 145, 456–465.
- Wisskirchen, K.; Kah, J.; Malo, A.; Asen, T.; Volz, T.; Allweiss, L.; Wettengel, J.M.; Lütgehetmann, M.; Urban, S.; Bauer, T.; et al. T cell receptor grafting allows virological control of hepatitis B virus infection. J. Clin. Investig. 2019, 129, 2932–2945.
- Ko, C.; Chakraborty, A.; Chou, W.-M.; Hasreiter, J.; Wettengel, J.M.; Stadler, D.; Bester, R.; Asen, T.; Zhang, K.; Wisskirchen, K.; et al. Hepatitis B virus genome recycling and de novo secondary infection events maintain stable cccDNA levels. J. Hepatol. 2018, 69, 1231–1241.
- Chong, C.L.; Chen, M.L.; Wu, Y.C.; Tsai, K.N.; Huang, C.C.; Hu, C.P.; Jeng, K.S.; Chou, Y.C.; Chang, C. Dynamics of HBV cccDNA expression and tran-scription in different cell growth phase. J. Biomed. Sci. 2011, 18, 96.
- Tu, T.; Zehnder, B.; Wettengel, J.M.; Zhang, H.; Coulter, S.; Ho, V.; Douglas, M.W.; Protzer, U.; George, J.; Urban, S. Mitosis of hepatitis B virus-infected cells in vitro results in uninfected daughter cells. JHEP Rep. 2022, 4, 100514.
- Li, M.; Sohn, J.A.; Seeger, C. Distribution of Hepatitis B Virus Nuclear DNA. J. Virol. 2018, 92, e01391-17.
- Dandri, M.; Burda, M.R.; Will, H.; Petersen, J. Increased hepatocyte turnover and inhibition of woodchuck hepatitis B virus repli-cation by adefovir in vitro do not lead to reduction of the closed circular DNA. Hepatology 2000, 32, 139–146.
- Lutgehetmann, M.; Volz, T.; Köpke, A.; Broja, T.; Tigges, E.; Lohse, A.W.; Fuchs, E.; Murray, J.M.; Petersen, J.; Dandri, M. In vivo proliferation of hepadnavirus-infected hepatocytes induces loss of covalently closed circular DNA in mice. Hepatology 2010, 52, 16–24.
- Allweiss, L.; Volz, T.; Giersch, K.; Kah, J.; Raffa, G.; Petersen, J.; Lohse, A.W.; Beninati, C.; Pollicino, T.; Urban, S.; et al. Proliferation of primary human hepatocytes and prevention of hepatitis B virus reinfection efficiently deplete nuclear cccDNA in vivo. Gut 2018, 67, 542–552.
- Macdonald, R.A. “Lifespan” of liver cells. Autoradio-graphic study using tritiated thymidine in normal, cirrhotic, and partially hepatectomized rats. Arch. Intern. Med. 1961, 107, 335–343.
- Magami, Y.; Azuma, T.; Inokuchi, H.; Kokuno, S.; Moriyasu, F.; Kawai, K.; Hattori, T. Cell proliferation and renewal of normal hepatocytes and bile duct cells in adult mouse liver. Liver Int. 2002, 22, 419–425.
- He, L.; Pu, W.; Liu, X.; Zhang, Z.; Han, M.; Li, Y.; Huang, X.; Han, X.; Li, Y.; Liu, K.; et al. Proliferation tracing reveals regional hepatocyte generation in liver homeostasis and repair. Science 2021, 371, eabc4346.
- Wei, Y.; Wang, Y.G.; Jia, Y.; Li, L.; Yoon, J.; Zhang, S.; Wang, Z.; Zhang, Y.; Zhu, M.; Sharma, T.; et al. Liver homeostasis is maintained by midlobular zone 2 hepatocytes. Science 2021, 371, eabb1625.
- Celis, J.E.; Celis, A. Cell cycle-dependent variations in the distribution of the nuclear protein cyclin proliferating cell nuclear antigen in cultured cells: Subdivision of S phase. Proc. Natl. Acad. Sci. USA 1985, 82, 3262–3266.
- Bravo, R.; Macdonald-Bravo, H. Changes in the nuclear distribution of cyclin (PCNA) but not its synthesis depend on DNA replication. EMBO J. 1985, 4, 655–661.
- Bravo, R.; Macdonald-Bravo, H. Existence of two populations of cyclin/proliferating cell nuclear antigen during the cell cycle: Association with DNA replication sites. J. Cell Biol. 1987, 105, 1549–1554.
- Mancini, R.; Marucci, L.; Benedetti, A.; Jezequel, A.-M.; Orlandi, F. Immunohistochemical analysis of S-phase cells in normal human and rat liver by PC10 monoclonal antibody. Liver Int. 1994, 14, 57–64.
- Nakajima, T.; Kagawa, K.; Ueda, K.; Ohkawara, T.; Kimura, H.; Kakusui, M.; Deguchi, T.; Okanoue, T.; Kashima, K.; Ashihara, T. Evaluation of hepatic proliferative activity in chronic liver diseases and hepatocellular carcinomas by proliferating cell nuclear antigen (PCNA) immunohisto-chemical staining of methanol-fixed tissues. J. Gastroenterol. 1994, 29, 450–454.
- Mason, W.S.; Jilbert, A.R.; Litwin, S. Hepatitis B Virus DNA Integration and Clonal Expansion of Hepatocytes in the Chronically Infected Liver. Viruses 2021, 13, 210.
More