Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Annalisa Marcuzzi and Version 2 by Conner Chen.
Neurodegenerative diseases comprise a wide spectrum of pathologies characterized by progressive loss of neuronal functions and structures. Despite having different genetic backgrounds and etiology, many studies have highlighted a point of convergence in the mechanisms leading to neurodegeneration: mitochondrial dysfunction and oxidative stress have been observed in different pathologies, and their detrimental effects on neurons contribute to the exacerbation of the pathological phenotype at various degrees.
- neuroinflammation
- mitochondria
- mitophagy
1. Introduction
Neurodegenerative diseases elicit the progressive degeneration or death of neural cells, provoking either movement disorders, such as in ataxias, Amyotrophic Lateral Sclerosis (ALS) and Multiple Sclerosis (MS), or cognitive impairments (dementias), such as Alzheimer’s Disease (AD) and Parkinson’s Disease (PD). These are pathologies with increasing incidence in developed countries, accompanying a progressive aging of the population [1][2][3][1,2,3].
In this context, Stanga et al. [4] determined that mitochondrial dysfunctions represent a crucial point of convergence in the pathogenesis of such neurodegenerative pathologies.
Recent studies, indeed, have highlighted the role of mitochondria not only in normal brain functions but also in the pathogenesis of its diseases: the main actors in these conditions are mitochondrial dysfunction and excessive oxidative stress, which are often found as linked events [5][6][5,6]. Neuronal transmission, integrity and survival all depend on efficient mitochondria, responsible for ATP production (required for the transport and release of neurotransmitters) and contributing to calcium homeostasis and apoptosis control [5][7][5,7]. At the same time, the oxidative phosphorylation leading to ATP generation also produces free radicals, namely reactive oxygen species (ROS), that make mitochondria susceptible to oxidative stress. Therefore, even minor alterations of the redox equilibrium induce ROS accumulation and mitochondrial dysfunction, accompanied by severe consequences, primarily energetic failure, excitotoxicity and rupture of the mitochondrial membrane [5][7][8][5,7,8].
2. Mitochondrial Dynamics
However, mitochondria are extremely plastic organelles, and the endogenous antioxidant system (involving glutathione and other reducing agents) is not the only coping mechanism that a cell can use (Figure 1).
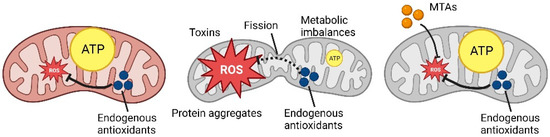
Figure 1. Schematic representation of ROS production and MTAs (mitochondria-targeted antioxidants) mechanism (Created with
Schematic representation of ROS production and MTAs mechanism (Created with
).
Mitochondria’s structure can be rearranged through two important processes [5][9][5,9]: mitochondrial fission, the division of a mitochondrion into two separate mitochondria, an event that generates a fragmented mitochondrial network with several rounded mitochondria; contrarily, mitochondria fusion is the merging of two or more mitochondria that generates a hyperfused network with elongated mitochondria highly connected with one another. Mitochondrial dynamics—that is, the balance between the fission and fusion mechanisms—represents the pivotal point in mitochondrial and, more broadly, cellular homeostasis [9][10][9,10]. It can be considered that the main purpose of this equilibrium is not limited to the maintenance of mitochondrial function; it is also crucial to guarantee an adequate energetic and metabolic supply to the cell [11][12][11,12]. Morphologic changes at this level represent a cellular response to specific physio-pathologic conditions that require a considerable energetic consumption from the very same mitochondria [13][14][13,14].
In non-physiologic conditions, an external stimulus elicits an adaptive response that translates into the remodeling of mitochondria, which may induce a partial or complete blockade of their motility or, on the other hand, the fragmentation of their network. Specifically, this last event is characterized by a condition of high ROS production that determines oxidative stress and cell death, in turn triggering an increased mitochondrial fission [15][16][15,16]. In this scenario, the fragmentation might be useful to prevent the damage caused by ROS and to facilitate the elimination of damaged mitochondria. On the contrary, an excessive mitochondrial fusion seems to be associated with a pronounced mitochondrial elongation that confers protection from the autophagy mechanism, inhibiting the formation of phagosomes [17][18][17,18].
The process of mitochondria degradation, called mitophagy, is finely regulated [19][20][21][22][19,20,21,22]: PINK1 (PTEN Induced Kinase 1 or PARK6), a protein kinase, translocates on the outer membrane of a damaged mitochondrion that loses physiological membrane potential. Parkin (or PARK2), a ubiquitin ligase, is recruited and activated by PINK1, leading to ubiquitination of the mitochondrion, which, in this way, is engulfed by a phagosome and degraded when the phagosome fuses with a lysosome. This process is activated to get rid of fragmented mitochondria and recycle their components [20][23][20,23], destined to be the biogenesis of new functional mitochondria, and is particularly critical for highly demanding cells such as neurons that need to keep under control the energy supply and redox status, thus relying on the efficient elimination and replacement of dysfunctional mitochondria with a high ROS content [24][25][26][24,25,26].
Generally speaking, ROS mostly derive from the mechanism of energy production taking place in the mitochondria, and they are countered by the cellular antioxidant system [27][28][27,28]. Indeed, the oxidative stress is represented by an imbalance between the concentration of generated ROS and the antioxidant potential that counters them, determining an alteration of the intracellular signaling and redox status. It can be observed a wide spectrum of adaptive cellular responses as the oxidative stress increases: in fact, a low level determines changes in genes and protein expression with no structural changes in the mitochondria, while a significant level can lead to high fragmentation and eventually cell death [29][30][29,30].
3. Neurodegenerative Disorders Characterized by Mitochondrial Involvement
In the past 30 years, an increasing body of evidence has pointed at the role of mitochondria in the brain homeostasis, as well as the pathogenesis of neurodegenerative diseases, in which they may have a main role or be involved in a second moment exacerbating the neuronal damage [31] (Table 1).
Table 1.
Mechanisms involved in neurodegenerative diseases.
| Neurodegenerative Diseases | Mechanisms Involved | References |
|---|---|---|
| Parkinson’s disease (PD) |
|
[32][33]33[34][35][36][37][38][39][40],34[,3541,36][,3742][32,,38,39,40,41,42] |
| Alzheimer’s disease (AD) |
|
[32][34][43][44][45][46][32,34,43,44,45,46] |
| Amyotrophic lateral sclerosis (ALS) |
|
[47]][47[48],48[49][50,49,50] |
| Multiple Sclerosis (MS) |
|
[51][52][53][54][55][51,52,53,54,55] |
| Huntington’s disease (HD) |
|
[19][56][57][58][59][60][61][62][63][19,56,57,58,59,60,61,62,63] |
Parkinson’s Disease is a progressive motor disorder, affecting 1% of the world population over the age of 60 years, caused by neurodegeneration occurring in the substantia nigra [32][33][34][32,33,34]. Symptoms comprise the characteristic resting tremor and postural instability, muscle rigidity, bradykinesia and, in some cases, sleeping disorders and cognitive impairment. The nigral dopaminergic neurons are the major cell type involved in the disease, due to their high reliance on oxidative phosphorylation, determining their susceptibility to oxidative stress [32][33][32,33]. Moreover, in PD patients, this brain region shows an accumulation of intraneuronal Lewy bodies that retain aggregated proteins, mainly α-synuclein but also neurofilaments, ubiquitin and others [33][34][35][33,34,35]. Physiologically, α-synuclein seems to be implicated in the transport and release of neurotransmitters; however, when aggregates are formed, they can interact with transport machineries present on the outer mitochondrial membrane, blocking their activity, with negative consequences on mitochondrial functions, primarily oxidative phosphorylation, followed by ROS accumulation, alteration of the membrane potential and impaired calcium homeostasis [33][34][33,34]. This process was confirmed both in vivo, where mutant α-synuclein potentiated the effects of PD-inducing toxins in a mouse model [36], and in patients’ biopsies [37]. Moreover, α-synuclein can be released in the extracellular space and taken up by nearby neurons, where it induces protein aggregation, spreading the formation of Lewy bodies. Other evidence shows a correlation between mitochondrial dysfunctions and α-synuclein aggregation: cases of MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) intoxication [38] and studies on animals exposed to MPTP [39] and herbicides (paraquat and rotenone) [40][41][42][40,41,42] reported the inhibition of Complex I of the respiratory chain, with the consequent accumulation of ROS and aggregates, even if the process leading to the latter effect still needs in-depth studies. Cases of familial PD, instead, are linked to alterations in the process of mitophagy: mutations in the gene PINK1 and Parkin were identified as drivers of PD, since the impaired degradation of dysfunctional mitochondria leads to neurodegeneration [33][34][33,34].
Alzheimer’s Disease is an age-related neurological disease, caused by the progressive degeneration of cognitive functions, starting with memory loss up to the loss of ability to perform daily tasks. These problems are caused by the progressive accumulation of amyloid β (Aβ) both in the extracellular milieu and within the cell and the formation of hyperphosphorylated tau-protein tangles [32][34][43][44][32,34,43,44]. The link between Aβ aggregation and mitochondrial dysfunction has been demonstrated, and some studies have revealed a bidirectional relationship. It was proposed that mitochondrial dysfunction and altered metabolism were early features of AD, a hypothesis supported by different studies [32][34][43][45][46][32,34,43,45,46] revealing alterations of the metabolism independent of Aβ that caused the formation of Aβ aggregates and tau fibrils, the latter being a consequence of tau protein hyperphosphorylation. Mitochondrial defects and oxidative stress may result from age, sedentary lifestyle, poor exercise and a high-calorie diet [43][45][43,45]. At the same time, in other studies, researchers observed that exposure to Aβ aggregates was the driving force of mitochondrial damage, impaired calcium homeostasis, ROS overproduction, oxidative stress and energy shortage. Fibrils of tau protein also contributed to respiratory chain impairment [32][34][43][44][45][32,34,43,44,45]. Ultimately, both Aβ and tau aggregates interfere with mitochondria trafficking, blocking their transport towards synapses; mitochondrial dynamics and biogenesis, shifting the equilibrium towards fission and generating small fragmented mitochondria that cannot be replaced by new mitochondria; and mitophagy, blocking the signaling molecules mediating the fusion with autophagosomes/lysosomes [32][34][43][44][45][46][32,34,43,44,45,46].
Amyotrophic Lateral Sclerosis is characterized by progressive muscle paralysis mediated by damage to the upper and lower motor neurons, causing severe debilitation and later death within a few years after the diagnosis [47][48][49][50][47,48,49,50]. Genetic mutations responsible for the pathogenesis are still under investigation, with few confirmed as causative mutations. Despite the genetic variability, the common hallmarks of ALS are the mitochondrial and endoplasmic reticulum abnormalities, as well as the disruption of the signaling between these compartments [47][48][49][50][47,48,49,50]. Mitochondria dysfunctions are caused by mutated proteins that can either form aggregates or stimulate mitochondria fragmentation. Another characteristic of ALS is the presence of aggregated, swollen and vacuolated mitochondria [47][48][49][50][47,48,49,50]. Therefore, mitochondria become dysfunctional, showing impaired ATP production and ROS removal, leading to progressive neuron death, which exacerbates the pathological condition.
Multiple Sclerosis is an autoimmune disease that causes muscular weakness, ataxia, cognitive impairments and autosomal nervous system dysfunction. The main driving force is the autoreactivity of CD4+ T-helper type 1 cells against myelin components [51][52][53][51,52,53]. When immune cells cross the blood–brain barrier and reach the CNS, they provoke demyelination and inflammation, with profound deleterious effects on neurons. Myelin sheath loss hampers the action potential, because the exposure of more axonal membrane portions slows down the signal transmission and increases the energetic demand to activate the membrane ion channels [54]. Consequently, the mitochondrial respiratory chain is overactivated to supply sufficient ATP, causing the accumulation of ROS and exhaustion of antioxidant systems, with the contribution of tissue inflammation [51][52][53][55][51,52,53,55]. ROS are responsible for damages to mitochondrial membranes and mtDNA, lipid peroxidation and mitochondrial fragmentation, impairing the already worn out electron transport chain, with consequent decreased ATP production, which worsens the action potential transmission [51][52][53][55][51,52,53,55].
Huntington’s Disease is an autosomal-dominant disease involving progressive neurodegeneration that affects 5–10 per 100,000 individuals in Western countries. The genetic basis of this disease is a copy number variation of a CAG (cytosine–adenine–guanine) repeat in the huntingtin gene (HTT), causing expansion of the repeats that generates a mutant HTT protein with a longer N-terminal polyglutamine sequence [19][56][57][58][19,56,57,58]. Affected neurons are mostly found in the striatum, causing tissue damage and atrophy; however, the damage can spread to cortical areas and other regions. HTT mutation leads to protein accumulation and aggregation in the cytoplasm and nucleus, where it interferes with other proteins, impairing normal neuronal functions [19][58][19,58]. Normally, the protein participates in different trafficking mechanisms, including axonal transport, nuclear transport, autophagy and mitophagy, in which it interacts with and stabilizes different protein complexes [19][58][19,58]. Alterations of its activity lead to oxidative stress, mitochondrial dysfunction and excitotoxicity. In fact, in the striatum of mouse models [59] and human biopsies [60][61][60,61] of HD, researchers found higher levels of ROS than in control subjects, associated with reduced or no activity of Complexes II, III and IV of the respiratory chain. HTT aggregates also induce increased mitochondrial fission, reducing the energetic efficiency with a consequent ROS overproduction [56][62][56,62]. This is exacerbated by impaired mitochondria trafficking and mitophagy, which cause the accumulation of dysfunctional mitochondria at synapses that, in turn, start degenerating because of the lack of ATP and neurotransmitter release [19][56][63][19,56,63].