Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Won Wong and Version 2 by Conner Chen.
ZIKV belongs to the genus of Flavivirus in the Flaviviridae family that comprises multiple deadly human pathogens, including the dengue virus (DENV), Japanese encephalitis (JEV), the yellow fever virus (YFV), and the West Nile virus (WNV). ZIKV infection is known to result in severe manifestations including neurological complications in adults and congenital abnormalities in newborns.
- Zika virus
- neurological symptoms
- gene and structure
1. Gene and Structure
The zikaZIKV virus genome is made up of 10.8 kb positive-sense, single-stranded RNA flanked by the 5′ and 3′ untranslated regions (UTRs) with a single open reading frame (ORF) [1][2][22,23]. The ORF region encodes a single polypeptide, which is processed into three structural proteins, including a capsid (C), precursor membrane (prM), and envelope (E), as well as seven non-structural proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5). The C proteins construct the icosahedral viral capsid, which encapsulates the viral genomic RNA, while the prM and E proteins are anchored on the outer membrane. prM is cleaved by host cell furin protease to generate mature virion, whereas the E protein is involved in binding and membrane fusion, which permit viral entry into the host cells [3][24].
NS1 is a glycoprotein of approximately 60 kDa that serves as an RNA replication complex in flaviviruses. Due to the importance of its replicative function in flaviviruses, the NS1 sequence is highly conserved, whereby ZIKV shares a >50% sequence homology with DENV2 ( dengue virus (DENV)) and West Nile virus (WNV)NV NS1 [4][25]. The protein is found in the following different forms in various locations in the host cells: (i) dimers in membrane-bound vesicles in the lumen of the endoplasmic reticulum, (ii) dimers in association with the membranes of flavivirus-infected cells, and (iii) highly immunogenic hexamers that are secreted into extracellular fluid [5][6][26,27].
NS2A is a 22 kDa transmembrane protein located in the endoplasmic reticulum, and it plays a critical role in the viral replication process [7][28]. It also interacts with NS2B and NS3 to recruit viral RNA, prM, and E to the virion assembly site for virus morphogenesis [8][9][29,30]. It has been suggested that the NS2A protein participates in ZIKV-induced neurological damage, as it interacted with multiple adherent junctions in an embryonic mouse cortex and impaired radial glial cell proliferation in human forebrain organoids [10][31].
NS2B/NS3 forms the viral protease complex that is involved in genome replication and cleavage of the viral polypeptide [11][32]. NS3 carries the protease domain at the N-terminus and the RNA helicase domain at the C-terminus, while NS2B acts as the membrane-bound domain that positions NS3 to its substrate and forms part of the NS3 catalytic domain for substrate binding [12][13][33,34].
NS4A/NS4B cause neurological impairment via manipulating the cellular survival and autophagy signaling pathways [14][35]. The introduction of NS4A or NS4B in human fetal neural stem cells (NSCs) resulted in impaired neurosphere formation, likely through inhibiting Akt kinase phosphorylation at Thr308 and Ser473 and mammalian target of rapamycin (mTOR) phosphorylation at Ser2448, which disrupted autophagy [15][36]. NS5, on the other hand, comprises methyltransferase with a short linker to the RNA-dependent RNA polymerase (RdRP) that is vital for RNA replication. It performs guanylyl transferase activity to catalyze the de novo formation of a methylated RNA cap structure using a triphosphorylated RNA transcript [16][37].
2. African and Asian ZIKV Lineages
Phylogenetic analysis has classified the ZIKV into two major genotypes, namely, the African and Asian lineages; the latter is further subdivided into the local Asian or contemporary American subclades [17][18][38,39]. The African and Asian ZIKV lineages display differences in virulence, transmissibility, and replication kinetics [19][20][21][40,41,42], despite sharing a high degree of similarity (>88.9%) in their genomic sequences [1][22]. The African ZIKV strain demonstrates a higher rate of transmissibility in the mosquito vector Aedes aegypti compared to its Asian counterpart [22][43]. Its infection results in a higher rate of lethality and can lead to cases of fetal death [23][44]. In contrast, the low-virulence Asian lineage does not induce early cell death, but it may lead to chronic infections in the fetal central nervous system [24][45]. The reemergence of ZIKV epidemics in 2015 were dominated by a strain of Asian ZIKV lineage that is commonly named the American strain [25][46]. Preceding the outbreak, ZIKV Asian lineage had been associated with an evolutionary mutation in the viral E gene (V473M) during replication and transmission between mosquito and host [26][47]. This mutation increases its virulence and viremia generation, hence enhancing transmission, which could be a critical determinant in the epidemics. Intriguingly, an effort to inverse the V473M substitution in the epidemic ZIKV strain isolated in Puerto Rico in 2015 reversed the pathogenic phenotypes of the virus [26][47]. Recent ZIKV outbreaks of the local Asian lineage have been reported in different states of India in 2018 and 2021 [27][28][29][48,49,50].
3. Transmission and Life Cycle
Similar to other flaviviruses, ZIKV is vector-borne and can be disseminated by infected female Ae. aegypti and Ae. albopictus mosquitoes. However, it differs from DENV in that it can be transmitted vertically from a pregnant mother to a baby [30][31][51,52], via blood transfusions [32][53], and via sexual intercourse [33][34][54,55]. Vertical transmission is observed in the mosquito vectors, Ae. aegypti and Ae. albopictus, to the larvae of infected mosquitoes [30][35][51,56].
The life cycle of ZIKV is highly similar to other members of the Flavivirus family; it begins with the entry of a viral particle into a host cell via clathrin-mediated endocytosis modulated by the binding of viral protein E. Viral entry is facilitated by the rolling and accumulation of viral particles along a host cell surface. The differential expression of various binding factors in a host cell surface dictates the viral tropism. The presence of the transmembrane receptor tyrosine kinase protein anexelekto (AXL), which is highly expressed by neural cells, dendritic cell-specific intracellular adhesion molecule 3-grabbing nonintegrin (DC-SIGN), tyrosine-protein kinase receptor (TYRO3,) and T-cell immunoglobulin and mucin domain 1 (TIM-1) on host cells is vital for the viral endocytic event [36][37][57,58].
When ZIKV reaches a clathrin-expressing surface, the host cell membrane invaginates and fuses with the viral membrane in the presence of acidic host cell cytoplasm, allowing the viral genome to be released into the cytoplasm (Figure 1). Following the release, protein translation occurs, and the newly synthesized viral proteins will be recruited into the endoplasmic reticulum for assembly [38][59]. With help from the NS proteins, new and immature viral particles migrate to the Golgi body, where precursor prM proteins are cleaved. Mature virions are subsequently released from the cell and are ready for a new cycle of infection. Occasionally, an immature viral particle carrying the uncleaved prM can be released [39][60].
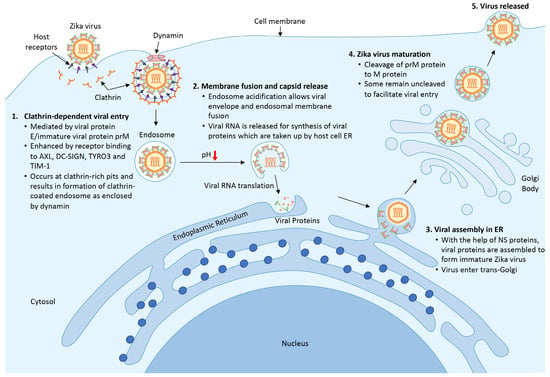
Figure 1. Life cycle of ZIKV. (1) ZIKV encounters host cells and binds to host receptors (AXL, DC-SIGN, TYRO3, and TIM-1) via viral protein E and prM and initiates clathrin-dependent viral entry. (2) Upon entering the cell, the endosome matures and acidifies, resulting in the release of viral RNA and the translational process to synthesize viral proteins. (3) New viral proteins are assembled into an immature viral particle within the endoplasmic reticulum. (4) Immature viral particles enter the trans-Golgi network where prM protein is cleaved into a mature virus. Finally, the newly formed virus is released to the surrounding areas and is ready for subsequent infection.
4. Symptoms Caused by ZIKV Infection
During the Yap Island outbreak in 2007, a majority of the cases were mild, with clinical symptoms that included low-grade fever, maculopapular rash, arthralgia, and conjunctivitis [40][61]. Severe neurological complications of ZIKV infection were observed in a small number of cases during the French Polynesian outbreak. This was highlighted by the increased prevalence of an autoimmune disease causing acute or subacute flaccid paralysis, known as Guillain–Barre syndrome, to approximately a 20-fold higher rate than was expected (1/2 in 100,000 people per year) in adults, approximately 3 weeks following the ZIKV outbreak [41][62]. Trends of microcephaly among newborns of infected mothers were reported during the outbreak in Brazil from 2015 to 2016 [42][63]. Other forms of neurological deficits, including meningoencephalitis [43][44][64,65], transverse myelitis [45][66], ophthalmic manifestation with optic nerve and retina complications [46][47][67,68], and other neuronal developmental defects [48][69], were identified among infants. Subsequent studies using human brain organoids [49][70], as well as animal models using macaques, mice, or fruit flies [31][50][51][52,71,72], have confirmed the viral neurotropism and developmental impact. Early neurological impairments, including severe intellectual disability, spastic tetraparesis, dysphagia, and failure to thrive [52][73], as well as severe motor impairment, were recently described in congenital ZIKV-infected children [53][74].
ZIKV causes neurological deficits through damaging neuronal development and proliferation. Li, et al. [54][15] showed that in human neural progenitor cells (NPCs), ZIKV infection caused cell-cycle arrest, apoptosis, and the inhibition of cell differentiation, which eventually gave rise to cortical thinning and microcephaly. Gabriel, et al. [55][10] reported that ZIKV infection resulted in the premature differentiation of NPCs, which was associated with centrosome perturbation, progenitor depletion, disrupted ventricular zone proliferation, impaired neurogenesis, and cortical thinning. In addition, Onorati, et al. [56][75] utilized a single-cell RNA-sequencing technique to investigate the effects of ZIKV on the neuropathogenesis of neocortical and spinal cord neuroepithelial stem cells, and they demonstrated that ZIKV infection caused disrupted cell mitoses, supernumerary centrosomes, structural disorganization, and cell death. Treatment with nucleoside analogs inhibited ZIKV replication and ZIKV-mediated death in neuroepithelial stem cells [56][75].