Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Bisera Stepanovska and Version 2 by Rita Xu.
Kidney cancer is among the top ten most common cancers to date. Within the kidney, renal cell carcinoma (RCC) is the most common solid lesion occurring. Mutations in the von Hippel–Lindau gene (Vhl) have attracted a lot of interest since this gene regulates the hypoxia inducible transcription factors HIF-1α and HIF-2α, which in turn drive the transcription of many genes that are important for renal cancer growth and progression, including genes involved in lipid metabolism and signaling.
- lipids
- sphingolipids
- glycosphingolipids
- free fatty acids
- kidney cancer
- renal cell carcinoma
1. Introduction
Kidney cancer is among the top ten most common cancers to date and accounts for about 3% of adult malignancies [1][2][1,2]. Malignant tumors can arise either from the renal parenchyma or the renal pelvis. In children, the most common kidney cancer is nephroblastoma (Wilms tumor), accounting for 1.1% of all kidney cancers [3], while in adults, renal cell carcinoma (RCC) is the most common neoplasm within the kidney. RCC originates from the renal epithelium, specifically from the proximal convoluted tubule and accounts for >90% of cancers in the kidney. The disease encompasses more than 10 histological and molecular subtypes, of which clear cell RCC (ccRCC) is the most common and accounts for most kidney cancer-related deaths [4][5][4,5]. It is characterized histologically by the accumulation of cholesterol esters, cholesterol, and other neutral lipids [6], which when dissolved during histological preparation methods show a clear cytoplasm. RCC can also be considered a metabolic disease because metabolic pathways are strongly altered in RCC, including glycolysis, amino acid metabolism, and lipid metabolism [7][8][7,8]
Understanding the biology of ccRCC starts with the discovery and characterization of the Vhl gene. The loss or mutation of the Vhl gene, at the short arm of chromosome 3, is generally considered to be one of the obligate initiating steps in the development of ccRCC [9]. Germline mutations of the Vhl gene cause autosomal dominant hereditary von Hippel –Lindau familial cancer syndrome characterized by an increased risk of tumor development in multiple organs, including the kidney [10]. Associated focal lesions, such as ccRCC, arise from the inactivation or silencing of the remaining normal (wild-type) Vhl allele. Remarkably, biallelic Vhl mutations or, less frequently, hypermethylation are very common in sporadic ccRCC, meaning that the Vhl gene behaves like a classical Knudson two-hit tumor suppressor gene. The main function of the Vhl gene product, pVHL, is to regulate the cell’s response to oxygen availability. It functions as a subunit of the E3 ubiquitin ligase complex, which mediates the proteasomal degradation of an oxygen-dependent transcription factor called hypoxia inducible factor (HIFα). HIFα exists as three isoforms, HIF-1α, HIF-2α, and HIF-3α, with the HIF-2α isoform being most directly associated with ccRCC carcinogenesis. Under hypoxic conditions, HIF-2α heterodimerizes with an aryl hydrocarbon receptor nuclear translocator (ARNT, also known as HIF-1β) to form an active transcription factor complex that upregulates the expression of hypoxia-inducible genes, such as vascular endothelial growth factor (VEGF) and erythropoietin (Epo), to counteract hypoxia and increase tissue oxygenation [11]. Under normal conditions, oxygen-dependent post-translational modifications on HIF-2α allow pVHL to recognize and target HIF-2α for rapid degradation. In RCC, the loss of pVHL thus mimics hypoxia and leads to excess HIF activity and the subsequent activation of the transcription of hundreds of HIF target genes that participate in angiogenesis, cell migration, epithelial–mesenchymal transition (EMT), extracellular matrix remodeling, glucose and lipid metabolism, immune evasion, and metastasis [12]. An important gene is the one encoding for VEGF which is a major driver of angiogenesis and thereby supplies the tumor with more nutrients and oxygen to accelerate its growth and progression. Drugs that inhibit VEGF production or its interaction with VEGF receptors have become a central approach of ccRCC treatment [13][14][13,14].
2. Sphingolipids in Renal Cancer
Sphingolipids represent a vast class of lipids characterized by the presence of a sphingoid backbone in their structure. They are important constituents of cellular membranes, but are increasingly acknowledged for their role as signaling molecules. Ceramide is the main hub of the sphingolipid pathways (Figure 1). It is produced either through the de novo synthetic pathway, the salvage pathway, or the hydrolytic pathway. Ceramide may then be phosphorylated to form ceramide 1-phosphate (C1P), deacylated to sphingosine, or condensed with phosphatidylcholine to give sphingomyelin or glucose/galactose to give cerebrosides [15][16][18,19]. Ceramide is known as a pro-apoptotic molecule, and many commonly used chemo-therapeutic agents induce cancer cell apoptosis by activating the acid sphingomyelinase and increasing ceramide formation [17][20]. Additionally, in RCC cells, exposure to exogenous C6-ceramide, or increasing endogenous ceramide by a ceramidase inhibitor, had a cytotoxic effect [18][21].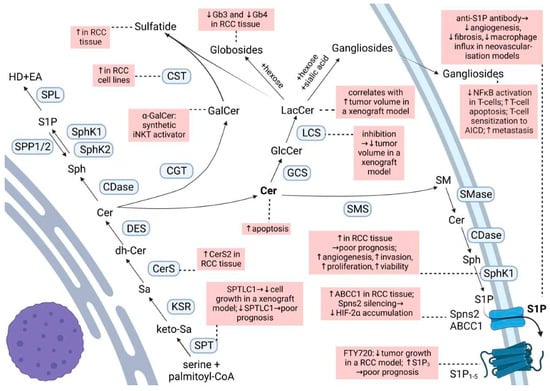
Figure 1. Sphingolipid biosynthesis and degradation routes. Changes reported for RCC are highlighted in pink boxes. For abbreviations, see text.
3. Glycosphingolipids in Renal Cancer
Galactosylceramide, or galactocerebroside, is produced from ceramide by the attachment of a galactose residue at the 1-hydroxyl moiety. α-Galactosylceramide (KRN-7000, α-GalCer) is a synthetic glycosphingolipid which acts as a synthetic iNKT (invariant Natural Killer T) cell ligand when presented by CD1d [45][48]. This interaction activates the iNKT and increases the number of iNKT and the production of pro-inflammatory cytokines which later activate the NK, tumor-specific, CD4+, CD8+ T cells, and B cells [46][49]. Numerous clinical trials have demonstrated tumor regression, a stable disease, partial response, or increased median survival time with α-GalCer therapy in various cancers; however, studies in RCC are missing [46][49]. Although this immunotherapeutic vaccine approach was suggested to be of benefit in RCC [45][48], the efficacy is unclear due to contradictory results and scarce studies. In this regard, in vitro α-GalCer-loaded dendritic cells induced the proliferation of iNKT cells derived from a pediatric papillary RCC [47][50]. However, NK T cells isolated from peripheral blood mononuclear cells (PBMCs) of a fraction of patients with metastatic (m)RCC showed no functional activity towards autologous tumor cells in the presence of α-GalCer [48][51]. Galactosylceramide is used by cerebroside/galactosylceramide–sulfotransferase (CST) to produce sulfatide. An elevated expression of sulfatide is commonly found in many human cancer cell lines and tissues and may possibly be used as a biomarker of some cancer cells [49][52]. Sulfatide is a major L-selectin ligand in the kidney, and the binding between L-selectin and sulfatide plays an essential role in monocyte infiltration into the kidney interstitium [49][52]. In various RCC cell lines, a marked increase of CST mRNA and activity was observed [49][52]. Moreover, lactosyl- and galactosylceramide sulfate are markedly increased in RCC as compared to healthy tissue, accompanied by significantly elevated activities of their respective sulfotransferases [50][51][53,54]. This is also reflected in the plasma and urine of RCC patients, where elevated concentrations of lactosylsulfatides were stage-dependent and more emphasized in late-stage RCC [52][55]. Nevertheless, Porubsky et al. (2021) could not confirm an association between CST expression and malignant clinical behavior of RCC [53][56]. Thus, the role of sulfoglycosphingolipids in RCC beyond the potential role as biomarkers for early RCC diagnosis [54][57] at this moment lacks evidence. The glucosylceramide synthase (GCS) is overexpressed in metastatic breast carcinoma [55][58] and drug-resistant breast, ovary, cervical, and colon cancer cells [56][59]. GCS upregulation is also part of the genetic signature for the progression and metastasis of RCC based on the results of gene-expression profiling of human RCC tumor samples [57][60]. Since an overexpression of GCS confers drug resistance and the suppression of GCS expression overcomes the resistance by enhancing drug uptake and ceramide-induced apoptosis in breast cancer cells [55][58][58,61], this suggests a mechanism that should also be considered in RCC. Glucosylceramide serves as a substrate for the lactosylceramide synthase to build lactosylceramide. In a xenograft mouse model of RCC a significant correlation between the increase in the mass of lactosylceramide and the tumor volume was detected, and inhibition of GCS and lactosyl-ceramide synthase activities led to a decrease in tumor volume [59][62]. Starting from lactosylceramide, globosides can be formed by the attachment of sugar residues, and gangliosides by the attachment of sugar residues and sialic acid. It is now generally accepted that gangliosides produced by cancer cells play a role in immune escape. In the context of RCC, it was demonstrated that explanted RCC tumors produce soluble gangliosides that inhibit the nuclear factor κB activation of co-cultured T cells [60][63], sensitize T cells to activation-induced cell death [61][64], or directly induce T-cell apoptosis by caspase activation [62][65]. For instance, RCC patients present with increased apoptotic T cells compared with T cells from healthy donors, and the majority of those apoptotic T cells were GM2(+) which they acquired from tumor shedding [63][66]. GM2 originating from RCC was also shown to promote T cell dysfunction by down-regulating cytokine production [64][67]. Not only do RCCs display increased levels of the gangliosides GD1a, GM1, and GM2 as compared with cells of the normal kidney [62][65][65,68], but they also synthesize disialogangliosides which seemingly promote the metastatic capabilities through a mechanism involving the formation of microembolisms [66][69]. Disialosyl globopentaosylceramide (DSGb5) is a dominant ganglioside isolated from RCC tissues [67][70] which binds to sialic acid-binding Ig-like lectin-7 (Siglec-7) expressed on natural killer (NK) cells, thereby inhibiting NK-cell cytotoxicity [68][71]. Higher DSGb5 expression exhibits greater migration potential in ACHN RCC cells and is correlated with metastasis in RCC patients [68][69][71,72]. Other gangliosides, such as GalNAc disialosyl lactotetraosylceramide [70][73] and monosialosyl galactosylgloboside (MSGG) [71][72][74,75], bring a higher risk of metastasis; however, the exact mechanisms are still not thoroughly investigated. Unlike gangliosides, the globosides globotriaosylceramide (Gb3) and globotetraosylceramide (Gb4) are markedly reduced in ccRCC tissue as compared to healthy renal tissue, and they decrease progressively with increasing malignancy grade [26][29]. There seems to be a connection between the ganglioside and globoside content in RCC cells driven by the action of the plasma membrane sialidase NEU3 [73][76]. NEU3 silencing in a human primary RCC cell line led to an increase in ganglioside content (e.g., GD1a, GM2, and GM3), and a decrease in the globoside Gb3 content [73][76]. Moreover, the production of ganglio-series gangliosides was enhanced to the detriment of globo-series gangliosides, particularly MSGG [73][76]. The effects of this silencing on RCC cell malignant phenotype and behavior were significant and involved drug resistance, invasive potential, and adhesion [73][76]. Nevertheless, other mechanisms could still play a role in these findings, as an increase of GM3 simultaneous with a decrease of MSGG in the human RCC cell line ACHN following brefeldin A treatment was marked by growth suppression and correlated to the pattern observed in RCC cases with a more favorable prognosis [74][77]. Considering the proposed functions of gangliosides in other tumors, such as binding to endothelial cells through carbohydrate–carbohydrate interactions, modulation of adhesion receptors, or the promotion of tumor-associated angiogenesis [66][69], this opens new avenues of research in the roles of gangliosides in RCC progression. Altogether, gangliosides expressed on RCC tumors may be important markers of tumor progression and target antigens for immunotherapy.4. Free Fatty Acids in Renal Cancer
4.1. Exogenous Uptake of Fatty Acids
Over the last decades, extensive studies have approached the effect of free fatty acids (FA), particularly of ω3-polyunsaturated fatty acids (PUFAs) on cancer cells, and many epidemiological studies support the idea of a correlation between dietary FA intake and the development of cancer [75][78]. Traditionally, saturated FAs have long been considered harmful, whereas plant monounsaturated FAs (MUFAs) such as oleic acid and ω-3 PUFAs were associated with a lower cancer mortality (Figure 2). However, systematic reviews reveal only weak epidemiological evidence for a clear protection by MUFAs and ω3-PUFAs [76][77][79,80]. Certain studies even concluded that certain MUFAs and PUFAs can promote cancer development [78][79][80][81][82][81,82,83,84,85].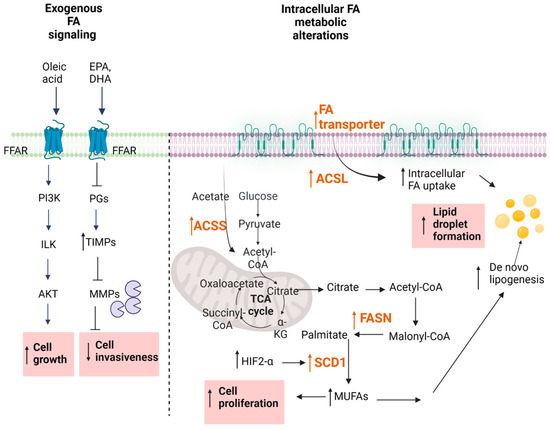
Figure 2. Fatty acid metabolism. Changes reported for RCC are highlighted in pink boxes. For abbreviations, see text.
4.2. Regulation of Fatty Acid Signaling in Cancer
The regulation of oncogenic signaling by FAs has also been considered as a novel therapeutic approach in RCC. Depending on the chain length, FAs can either freely enter the cell or use transport proteins [87][88][90,91] (Figure 2). Alternatively, cell surface FA receptors exist, denoted FFARs [89][90][92,93] which are subdivided into two groups, the long-chain FFARs (FFAR1/GPR40 and FFAR4/GPR120) and the short-chain FFARs (FFAR2/GPR43 and FFAR3/GPR41). All these receptors belong to the superfamily of GPCRs. Their involvement in cancer cell growth and progression are only now beginning to be unmasked, but it seems that the long-chain FFARs have a different role than short-chain FFARs [90][93]. Various in vitro studies in prostate, breast, ovarian, and colon cancer cells reveal that dual FFAR1/FFAR4 agonists can reduce the proliferation and migration of cancer cells, supporting the usefulness of these receptors as pharmacological targets [91][92][93][94][94,95,96,97]. However, so far, no reports are available for FFAR involvement in RCC. Clearly, it will be important to optimize such FFAR agonists for selectivity and potency when considering them for further development.4.3. Altered De Novo FA Synthesis
Metabolic reprogramming occurs because of mutations in cancer genes and alterations in cellular signaling. In addition to alterations in glucose and glutamine metabolism, increased de novo FA synthesis, uptake, and the suppression of FA oxidation, which eventually leads to lipid droplet (LD) formation, have been recently shown to be a hallmark of the disordered intermediary metabolism in cancer cells [95][96][98,99]. The fatty-acid synthase (FASN) is the key metabolic multi-enzyme that is responsible for the terminal catalytic step in FA synthesis (Figure 2). FASN is present at high levels in most human malignancies, especially in gynecological, prostate, and colon cancers [97][98][99][100,101,102], and it is correlates with a worse prognosis [100][103]. Therefore, FASN is speculated to be a new therapeutic target in RCC. In a first study, Horiguchi et al. showed increased FASN protein staining in immunohistochemical sections of RCC patients [101][104]. Positive FASN protein expression was associated with increased tumor aggressiveness and was an independent predictor of shortened cancer-specific survival, suggesting that FASN could be a predictive indicator of disease prognosis [101][104]. These data were later confirmed by another study [102][105], which assessed the differential mRNA expression of FASN in 533 ccRCC samples and 72 adjacent normal samples from a TCGA cohort. The data showed significantly increased FASN mRNA in ccRCC samples when compared to normal samples, and the elevated FASN mRNA correlated with a poor prognosis and malignant biological behaviors of ccRCC [102][105]. Similar data were also obtained by Yuan et al. [103][106] by using Western blot analysis and immunohistochemical staining of RCC tissue sections for FASN. FASN up-regulation and its association with a poor prognosis holds true for other cancer types as well [104][105][106][107,108,109], making this a universal cancer feature and thus supporting its usefulness as a therapeutic target of ccRCC. Wettersten et al. [107][110] revealed that in RCC, metabolic reprogramming is grade-dependent. Interestingly, they reported that the levels of shorter chain FFAs (6:0, 8:0, 9:0, 10:0, and 12:0) were decreased in a tumor grade-dependent manner and this was probably due to an increase in their utilization. Nevertheless, also in this study, FASN was found to be increased on a protein level in cancer tissue when compared to the adjacent nontumor tissue [108][111]. A functional analysis of FASN in human ccRCC cells showed that down-regulation or overexpression of FASN significantly regulates ccRCC cell proliferation and migration by regulating EMT. Moreover, FASN inhibition also increased the apoptotic rate, decreased lipid droplet formation, and suppressed the mRNA expression of hub genes in EMT [102][105]. On top of this, the pharmacological inhibition of FASN reduced the growth and invasiveness of renal cancer cells in vitro and in vivo. One possible mechanism could be the disturbance of cell membrane functioning by down-regulated Her2 and EGFR and downstream STAT3 signaling [101][104], which was shown to play an important role in pancreatic cancer metastasis [109][112]. The ability of FASN inhibition to suppress cancer cell growth was also proven in a cell line of a pediatric malignant rhabdoid kidney tumor [110][113]. Additionally, a proteomic analysis of tissue samples of a Wilms tumor confirmed that the expression of FASN was significantly increased in the tumor tissues as compared to adjacent tissues and this was associated with a poorer prognosis [111][112][114,115]. All these data suggest that FASN plays a key role in ccRCC carcinogenesis and that the FASN expression level could be equally used as a predictor of poor prognosis in both pediatric and adult renal tumors. Other than the de novo synthesis, other enzymes in the FA pathway are involved in the altered lipid metabolism of RCC such as altered FA activation, FA uptake, and the suppression of FA oxidation [96][99]. Once synthesized, FAs need to be activated by conversion to FA acyl-CoA esters by the action of acyl-CoA synthases (ACS) before they are further processed. Depending on the chain length of the Fas, ACSs are divided into different classes, comprising the very long chain (ACSVL), long chain (ACSL), medium chain (ACSM), and short chain (ACSS) synthases. Thus, ACSL converts FAs of C8-C22 chain lengths into an activated form. It represents a group of five isoforms, denoted ACSL1,3,4,5,6 [113][116]. Increased levels of all these enzymes have been suggested to be involved in molecular processes driving cancer growth and progression [113][114][116,117]. While a clear view on the relevance of all the ACSL isoforms in RCC is not yet available, it seems that at least ACSL3 could serve as a potential prognostic biomarker for immune infiltration in ccRCC [115][118]. Furthermore, ccRCC cells in vitro depend on ACSL3 for lipid droplet formation. Selective pharmacological inhibition or the genetic suppression of ACSL3 is cytotoxic for RCC cells and also reduces the tumor size in an orthotopic mouse cancer model [116][119]. These data propose that ACSL3 could indeed be a possible pharmacological target for RCC therapy.5. Conclusions and Perspectives
The reprograming of lipid metabolism is a typical characteristic feature of many tumors. Many of the so-far-described bioactive lipids can interfere with cancer-relevant molecular processes including cell proliferation and migration, apoptosis or survival, angiogenesis, and metastasis formation. Therefore, their involvement in RCC is very obvious and this has been approached in multiple in vitro and in vivo studies over the last decades. Another hallmark of RCC is the up-regulation of the HIF/VEGF signaling axis. It turns out that the targeting of the HIF/VEGF axis is very efficient in RCC treatment. However, resistance development is a main problem, which stresses the need for better treatment options. Not surprisingly, many of the bioactive lipids are regulating HIF/VEGF signaling or the opposite, are themselves regulated by HIF/VEGF, which highlights the attractivity of the lipids as a new targeting strategy for RCC, and it will be exciting to see whether novel therapeutics can arise from these lipid cascades.References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Huang, G.; Wei, W.; Liu, J. Molecular Imaging of Renal Cell Carcinoma in Precision Medicine. Mol. Pharm. 2022, 19, 3457–3470. [Google Scholar] [CrossRef]
- Chow, W.H.; Dong, L.M.; Devesa, S.S. Epidemiology and risk factors for kidney cancer. Nat. Rev. Urol. 2010, 7, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Moch, H.; Cubilla, A.L.; Humphrey, P.A.; Reuter, V.E.; Ulbright, T.M. The 2016 WHO Classification of Tumours of the Urinary System and Male Genital Organs-Part A: Renal, Penile, and Testicular Tumours. Eur. Urol. 2016, 70, 93–105. [Google Scholar] [CrossRef]
- Capitanio, U.; Bensalah, K.; Bex, A.; Boorjian, S.A.; Bray, F.; Coleman, J.; Gore, J.L.; Sun, M.; Wood, C.; Russo, P. Epidemiology of Renal Cell Carcinoma. Eur. Urol. 2019, 75, 74–84. [Google Scholar] [CrossRef]
- Drabkin, H.A.; Gemmill, R.M. Cholesterol and the development of clear-cell renal carcinoma. Curr. Opin. Pharmacol. 2012, 12, 742–750. [Google Scholar] [CrossRef]
- di Meo, N.A.; Lasorsa, F.; Rutigliano, M.; Loizzo, D.; Ferro, M.; Stella, A.; Bizzoca, C.; Vincenti, L.; Pandolfo, S.D.; Autorino, R.; et al. Renal Cell Carcinoma as a Metabolic Disease: An Update on Main Pathways, Potential Biomarkers, and Therapeutic Targets. Int. J. Mol. Sci. 2022, 23, 14360. [Google Scholar] [CrossRef] [PubMed]
- Lucarelli, G.; Loizzo, D.; Franzin, R.; Battaglia, S.; Ferro, M.; Cantiello, F.; Castellano, G.; Bettocchi, C.; Ditonno, P.; Battaglia, M. Metabolomic insights into pathophysiological mechanisms and biomarker discovery in clear cell renal cell carcinoma. Expert. Rev. Mol. Diagn. 2019, 19, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Latif, F.; Tory, K.; Gnarra, J.; Yao, M.; Duh, F.M.; Orcutt, M.L.; Stackhouse, T.; Kuzmin, I.; Modi, W.; Geil, L.; et al. Identification of the von Hippel-Lindau disease tumor suppressor gene. Science 1993, 260, 1317–1320. [Google Scholar] [CrossRef]
- Gnarra, J.R.; Tory, K.; Weng, Y.; Schmidt, L.; Wei, M.H.; Li, H.; Latif, F.; Liu, S.; Chen, F.; Duh, F.M.; et al. Mutations of the VHL tumour suppressor gene in renal carcinoma. Nat. Genet. 1994, 7, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Haase, V.H. Regulation of erythropoiesis by hypoxia-inducible factors. Blood Rev. 2013, 27, 41–53. [Google Scholar] [CrossRef]
- Wicks, E.E.; Semenza, G.L. Hypoxia-inducible factors: Cancer progression and clinical translation. J. Clin. Investig. 2022, 132, e159839. [Google Scholar] [CrossRef]
- Patel, S.A.; Nilsson, M.B.; Le, X.; Cascone, T.; Jain, R.K.; Heymach, J.V. Molecular Mechanisms and Future Implications of VEGF/VEGFR in Cancer Therapy. Clin. Cancer Res. 2023, 29, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Choueiri, T.K.; Kaelin, W.G., Jr. Targeting the HIF2-VEGF axis in renal cell carcinoma. Nat. Med. 2020, 26, 1519–1530. [Google Scholar] [CrossRef] [PubMed]
- Schirrmacher, V. From chemotherapy to biological therapy: A review of novel concepts to reduce the side effects of systemic cancer treatment (Review). Int. J. Oncol. 2019, 54, 407–419. [Google Scholar]
- Wallace, E.M.; Rizzi, J.P.; Han, G.; Wehn, P.M.; Cao, Z.; Du, X.; Cheng, T.; Czerwinski, R.M.; Dixon, D.D.; Goggin, B.S.; et al. A Small-Molecule Antagonist of HIF2alpha Is Efficacious in Preclinical Models of Renal Cell Carcinoma. Cancer Res. 2016, 76, 5491–5500. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Regulation of mammalian O2 homeostasis by hypoxia-inducible factor 1. Annu. Rev. Cell Dev. Biol. 1999, 15, 551–578. [Google Scholar] [CrossRef]
- Huwiler, A.; Kolter, T.; Pfeilschifter, J.; Sandhoff, K. Physiology and pathophysiology of sphingolipid metabolism and signaling. Biochim. Biophys. Acta 2000, 1485, 63–99. [Google Scholar] [CrossRef]
- Huwiler, A.; Pfeilschifter, J. Sphingolipid signaling in renal fibrosis. Matrix Biol. 2018, 68–69, 230–247. [Google Scholar] [CrossRef]
- Henry, B.; Moller, C.; Dimanche-Boitrel, M.T.; Gulbins, E.; Becker, K.A. Targeting the ceramide system in cancer. Cancer Lett. 2013, 332, 286–294. [Google Scholar] [CrossRef]
- Kim, Y.J.; Kim, E.A.; Sohn, U.D.; Yim, C.B.; Im, C. Cytotoxic Activity and Structure Activity Relationship of Ceramide Analogues in Caki-2 and HL-60 Cells. Korean J. Physiol. Pharmacol. 2010, 14, 441–447. [Google Scholar] [CrossRef][Green Version]
- Stepanovska, B.; Huwiler, A. Targeting the S1P receptor signaling pathways as a promising approach for treatment of autoimmune and inflammatory diseases. Pharmacol. Res. 2020, 154, 104170. [Google Scholar] [CrossRef]
- Takuwa, Y.; Du, W.; Qi, X.; Okamoto, Y.; Takuwa, N.; Yoshioka, K. Roles of sphingosine-1-phosphate signaling in angiogenesis. World J. Biol. Chem. 2010, 1, 298. [Google Scholar] [CrossRef]
- Hoefflin, R.; Harlander, S.; Abhari, B.A.; Peighambari, A.; Adlesic, M.; Seidel, P.; Zodel, K.; Haug, S.; Göcmen, B.; Li, Y. Therapeutic Effects of Inhibition of Sphingosine-1-Phosphate Signaling in HIF-2α Inhibitor-Resistant Clear Cell Renal Cell Carcinoma. Cancers 2021, 13, 4801. [Google Scholar] [CrossRef]
- Ader, I.; Brizuela, L.; Bouquerel, P.; Malavaud, B.; Cuvillier, O. Sphingosine kinase 1: A new modulator of hypoxia inducible factor 1α during hypoxia in human cancer cells. Cancer Res. 2008, 68, 8635–8642. [Google Scholar] [CrossRef] [PubMed]
- Bouquerel, P.; Gstalder, C.; Müller, D.; Laurent, J.; Brizuela, L.; Sabbadini, R.; Malavaud, B.; Pyronnet, S.; Martineau, Y.; Ader, I. Essential role for SphK1/S1P signaling to regulate hypoxia-inducible factor 2α expression and activity in cancer. Oncogenesis 2016, 5, e209. [Google Scholar] [CrossRef]
- Anelli, V.; Gault, C.R.; Cheng, A.B.; Obeid, L.M. Sphingosine kinase 1 is up-regulated during hypoxia in U87MG glioma cells: Role of hypoxia-inducible factors 1 and 2. J. Biol. Chem. 2008, 283, 3365–3375. [Google Scholar] [CrossRef] [PubMed]
- Salama, M.F.; Carroll, B.; Adada, M.; Pulkoski-Gross, M.; Hannun, Y.A.; Obeid, L.M. A novel role of sphingosine kinase-1 in the invasion and angiogenesis of VHL mutant clear cell renal cell carcinoma. FASEB J. 2015, 29, 2803. [Google Scholar] [CrossRef]
- Młynarczyk, G.; Mikłosz, A.; Suchański, J.; Reza, S.; Romanowicz, L.; Sobolewski, K.; Chabowski, A.; Baranowski, M. Grade-dependent changes in sphingolipid metabolism in clear cell renal cell carcinoma. J. Cell. Biochem. 2022, 123, 819–829. [Google Scholar] [CrossRef]
- Xu, Y.; Dong, B.; Wang, J.; Zhang, J.; Xue, W.; Huang, Y. Sphingosine kinase 1 overexpression contributes to sunitinib resistance in clear cell renal cell carcinoma. Oncoimmunology 2018, 7, e1502130. [Google Scholar] [CrossRef] [PubMed]
- Cuvillier, O.; Ader, I.; Bouquerel, P.; Brizuela, L.; Gstalder, C.; Malavaud, B. Hypoxia, therapeutic resistance, and sphingosine 1-phosphate. Adv. Cancer Res. 2013, 117, 117–141. [Google Scholar]
- Zhang, L.; Wang, X.; Bullock, A.J.; Callea, M.; Shah, H.; Song, J.; Moreno, K.; Visentin, B.; Deutschman, D.; Alsop, D.C. Anti-S1P Antibody as a Novel Therapeutic Strategy for VEGFR TKI-Resistant Renal CancerS1P Inhibition as a New Treatment for RCC. Clin. Cancer Res. 2015, 21, 1925–1934. [Google Scholar] [CrossRef][Green Version]
- Huwiler, A.; Bourquin, F.; Kotelevets, N.; Pastukhov, O.; Capitani, G.; Grütter, M.G.; Zangemeister-Wittke, U. A prokaryotic S1P lyase degrades extracellular S1P in vitro and in vivo: Implication for treating hyperproliferative disorders. PLoS ONE 2011, 6, e22436. [Google Scholar] [CrossRef]
- O’Brien, N.; Jones, S.T.; Williams, D.G.; Cunningham, H.B.; Moreno, K.; Visentin, B.; Gentile, A.; Vekich, J.; Shestowsky, W.; Hiraiwa, M. Production and characterization of monoclonal anti-sphingosine-1-phosphate antibodies 1. J. Lipid Res. 2009, 50, 2245–2257. [Google Scholar] [CrossRef] [PubMed]
- Caballero, S.; Swaney, J.; Moreno, K.; Afzal, A.; Kielczewski, J.; Stoller, G.; Cavalli, A.; Garland, W.; Hansen, G.; Sabbadini, R. Anti-sphingosine-1-phosphate monoclonal antibodies inhibit angiogenesis and sub-retinal fibrosis in a murine model of laser-induced choroidal neovascularization. Exp. Eye Res. 2009, 88, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Xie, B.; Shen, J.; Dong, A.; Rashid, A.; Stoller, G.; Campochiaro, P.A. Blockade of sphingosine-1-phosphate reduces macrophage influx and retinal and choroidal neovascularization. J. Cell. Physiol. 2009, 218, 192–198. [Google Scholar] [CrossRef]
- Pal, S.K.; Drabkin, H.A.; Reeves, J.A.; Hainsworth, J.D.; Hazel, S.E.; Paggiarino, D.A.; Wojciak, J.; Woodnutt, G.; Bhatt, R.S. A phase 2 study of the sphingosine-1-phosphate antibody sonepcizumab in patients with metastatic renal cell carcinoma. Cancer 2017, 123, 576–582. [Google Scholar] [CrossRef] [PubMed]
- Huwiler, A.; Zangemeister-Wittke, U. The sphingosine 1-phosphate receptor modulator fingolimod as a therapeutic agent: Recent findings and new perspectives. Pharmacol. Ther. 2018, 185, 34–49. [Google Scholar] [CrossRef] [PubMed]
- Fischl, A.S.; Wang, X.; Falcon, B.L.; Almonte-Baldonado, R.; Bodenmiller, D.; Evans, G.; Stewart, J.; Wilson, T.; Hipskind, P.; Manro, J. Inhibition of Sphingosine Phosphate Receptor 1 Signaling Enhances the Efficacy of VEGF Receptor InhibitionS1P1 Inhibition Improves VEGFR-Targeted Therapy. Mol. Cancer Ther. 2019, 18, 856–867. [Google Scholar] [CrossRef]
- Ying, Y.; Ma, X.; Fang, J.; Chen, S.; Wang, W.; Li, J.; Xie, H.; Wu, J.; Xie, B.; Liu, B. EGR2-mediated regulation of m6A reader IGF2BP proteins drive RCC tumorigenesis and metastasis via enhancing S1PR3 mRNA stabilization. Cell Death Dis. 2021, 12, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Bao, G.; Pei, J.; Cao, Y.; Zhang, C.; Zhao, P.; Zhang, Y.; Damirin, A. NF-κB and EGFR participate in S1PR3-mediated human renal cell carcinomas progression. Biochim. Et Biophys. Acta (BBA)-Mol. Basis Dis. 2022, 1868, 166401. [Google Scholar] [CrossRef]
- Glueck, M.; Koch, A.; Brunkhorst, R.; Ferreiros Bouzas, N.; Trautmann, S.; Schaefer, L.; Pfeilschifter, W.; Pfeilschifter, J.; Vutukuri, R. The atypical sphingosine 1-phosphate variant, d16: 1 S1P, mediates CTGF induction via S1P2 activation in renal cell carcinoma. FEBS J. 2022, 289, 5670–5681. [Google Scholar] [CrossRef]
- Hanada, K. Serine palmitoyltransferase, a key enzyme of sphingolipid metabolism. Biochim. Et Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2003, 1632, 16–30. [Google Scholar] [CrossRef]
- Zhu, W.K.; Xu, W.H.; Wang, J.; Huang, Y.Q.; Abudurexiti, M.; Qu, Y.Y.; Zhu, Y.P.; Zhang, H.L.; Ye, D.W. Decreased SPTLC1 expression predicts worse outcomes in ccRCC patients. J. Cell. Biochem. 2020, 121, 1552–1562. [Google Scholar] [CrossRef] [PubMed]
- Kong, Z.; Guo, X.; Zhao, Z.; Wu, W.; Luo, L.; Zhu, Z.; Yin, S.; Cai, C.; Wu, W.; Wang, D. SPTLC1 inhibits cell growth via modulating Akt/FOXO1 pathway in renal cell carcinoma cells. Biochem. Biophys. Res. Commun. 2019, 520, 1–7. [Google Scholar] [CrossRef]
- Wattenberg, B.W. The long and the short of ceramides. J. Biol. Chem. 2018, 293, 9922–9923. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.-K.; Maaß, M.; Quach, A.; Poscic, N.; Prangley, H.; Pallott, E.-C.; Kim, J.L.; Pierce, J.S.; Ogretmen, B.; Futerman, A.H. Dependence of ABCB1 transporter expression and function on distinct sphingolipids generated by ceramide synthases-2 and-6 in chemoresistant renal cancer. J. Biol. Chem. 2022, 298, 101492. [Google Scholar] [CrossRef]
- Schwaab, T.; Ernstoff, M.S. Therapeutic vaccines in renal cell carcinoma. Therapy 2011, 4, 369. [Google Scholar] [CrossRef]
- Companioni, O.; Mir, C.; Garcia-Mayea, Y.; LLeonart, M.E. Targeting Sphingolipids for Cancer Therapy. Front. Oncol. 2021, 11, 745092. [Google Scholar] [CrossRef]
- Lehmann, N.; Paret, C.; El Malki, K.; Russo, A.; Neu, M.A.; Wingerter, A.; Seidmann, L.; Foersch, S.; Ziegler, N.; Roth, L. Tumor Lipids of Pediatric Papillary Renal Cell Carcinoma Stimulate Unconventional T Cells. Front. Immunol. 2020, 11, 1819. [Google Scholar] [CrossRef]
- Vyth-Dreese, F.A.; Sein, J.; van de Kasteele, W.; Dellemijn, T.A.; van den Bogaard, C.; Nooijen, W.J.; de Gast, G.C.; Haanen, J.B.; Bex, A. Lack of anti-tumour reactivity despite enhanced numbers of circulating natural killer T cells in two patients with metastatic renal cell carcinoma. Clin. Exp. Immunol. 2010, 162, 447–459. [Google Scholar] [CrossRef]
- Takahashi, T.; Suzuki, T. Role of sulfatide in normal and pathological cells and tissues. J. Lipid Res. 2012, 53, 1437–1450. [Google Scholar] [CrossRef][Green Version]
- Sakakibara, N. Glycolipid alterations in human kidney carcinoma. [Hokkaido Igaku Zasshi] Hokkaido J. Med. Sci. 1989, 64, 75–82. [Google Scholar]
- Sakakibara, N.; Gasa, S.; Kamio, K.; Makita, A.; Koyanagi, T. Association of elevated sulfatides and sulfotransferase activities with human renal cell carcinoma. Cancer Res. 1989, 49, 335–339. [Google Scholar]
- Jirasko, R.; Idkowiak, J.; Wolrab, D.; Kvasnicka, A.; Friedecky, D.; Polanski, K.; Studentova, H.; Student, V.; Melichar, B.; Holcapek, M. Altered Plasma, Urine, and Tissue Profiles of Sulfatides and Sphingomyelins in Patients with Renal Cell Carcinoma. Cancers 2022, 14, 4622. [Google Scholar] [CrossRef] [PubMed]
- Porubsky, S.; Nientiedt, M.; Kriegmair, M.C.; Siemoneit, J.-H.H.; Sandhoff, R.; Jennemann, R.; Borgmann, H.; Gaiser, T.; Weis, C.-A.; Erben, P. The prognostic value of galactosylceramide-sulfotransferase (Gal3ST1) in human renal cell carcinoma. Sci. Rep. 2021, 11, 1–11. [Google Scholar] [CrossRef]
- Jirásko, R.; Holčapek, M.; Khalikova, M.; Vrána, D.; Študent, V.; Prouzová, Z.; Melichar, B. MALDI orbitrap mass spectrometry profiling of dysregulated sulfoglycosphingolipids in renal cell carcinoma tissues. J. Am. Soc. Mass Spectrom. 2017, 28, 1562–1574. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-Y.; Patwardhan, G.A.; Xie, P.; Gu, X.; Giuliano, A.E.; Cabot, M.C. Glucosylceramide synthase, a factor in modulating drug resistance, is overexpressed in metastatic breast carcinoma. Int. J. Oncol. 2011, 39, 425–431. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-Y.; Gupta, V.; Patwardhan, G.A.; Bhinge, K.; Zhao, Y.; Bao, J.; Mehendale, H.; Cabot, M.C.; Li, Y.-T.; Jazwinski, S.M. Glucosylceramide synthase upregulates MDR1 expression in the regulation of cancer drug resistance through cSrc and β-catenin signaling. Mol. Cancer 2010, 9, 1–15. [Google Scholar] [CrossRef]
- Jones, J.; Otu, H.; Spentzos, D.; Kolia, S.; Inan, M.; Beecken, W.D.; Fellbaum, C.; Gu, X.; Joseph, M.; Pantuck, A.J. Gene signatures of progression and metastasis in renal cell cancer. Clin. Cancer Res. 2005, 11, 5730–5739. [Google Scholar] [CrossRef]
- Liu, Y.-Y.; Han, T.Y.; Yu, J.Y.; Bitterman, A.; Le, A.; Giuliano, A.E.; Cabot, M.C. Oligonucleotides blocking glucosylceramide synthase expression selectively reverse drug resistance in cancer cells. J. Lipid Res. 2004, 45, 933–940. [Google Scholar] [CrossRef]
- Chatterjee, S.; Alsaeedi, N.; Hou, J.; Bandaru, V.V.R.; Wu, L.; Halushka, M.K.; Pili, R.; Ndikuyeze, G.; Haughey, N.J. Use of a glycolipid inhibitor to ameliorate renal cancer in a mouse model. PLoS ONE 2013, 8, e63726. [Google Scholar] [CrossRef] [PubMed]
- Uzzo, R.G.; Rayman, P.; Kolenko, V.; Clark, P.E.; Cathcart, M.K.; Bloom, T.; Novick, A.C.; Bukowski, R.M.; Hamilton, T.; Finke, J.H. Renal cell carcinoma–derived gangliosides suppress nuclear factor-κB activation in T cells. J. Clin. Investig. 1999, 104, 769–776. [Google Scholar] [CrossRef]
- Finke, J.H.; Rayman, P.; George, R.; Tannenbaum, C.S.; Kolenko, V.; Uzzo, R.; Novick, A.C.; Bukowski, R.M. Tumor-induced sensitivity to apoptosis in T cells from patients with renal cell carcinoma: Role of nuclear factor-κB suppression. Clin. Cancer Res. 2001, 7, 940s–946s. [Google Scholar]
- Kudo, D.; Rayman, P.; Horton, C.; Cathcart, M.K.; Bukowski, R.M.; Thornton, M.; Tannenbaum, C.; Finke, J.H. Gangliosides expressed by the renal cell carcinoma cell line SK-RC-45 are involved in tumor-induced apoptosis of T cells. Cancer Res. 2003, 63, 1676–1683. [Google Scholar]
- Biswas, S.; Biswas, K.; Richmond, A.; Ko, J.; Ghosh, S.; Simmons, M.; Rayman, P.; Rini, B.; Gill, I.; Tannenbaum, C.S. Elevated levels of select gangliosides in T cells from renal cell carcinoma patients is associated with T cell dysfunction. J. Immunol. 2009, 183, 5050–5058. [Google Scholar] [CrossRef][Green Version]
- Biswas, K.; Richmond, A.; Rayman, P.; Biswas, S.; Thornton, M.; Sa, G.; Das, T.; Zhang, R.; Chahlavi, A.; Tannenbaum, C.S. GM2 expression in renal cell carcinoma: Potential role in tumor-induced T-cell dysfunction. Cancer Res. 2006, 66, 6816–6825. [Google Scholar] [CrossRef]
- Hoon, D.S.; Okun, E.; Neuwirth, H.; Morton, D.L.; Irie, R.F. Aberrant expression of gangliosides in human renal cell carcinomas. J. Urol. 1993, 150, 2013–2018. [Google Scholar] [CrossRef] [PubMed]
- Ito, A.; Handa, K.; Withers, D.A.; Satoh, M.; Hakomori, S.-i. Binding specificity of siglec7 to disialogangliosides of renal cell carcinoma: Possible role of disialogangliosides in tumor progression. FEBS Lett. 2001, 504, 82–86. [Google Scholar] [CrossRef]
- Wu, D.-Y.; Adak, A.K.; Kuo, Y.-T.; Shen, Y.-J.; Li, P.-J.; Hwu, J.R.; Lin, C.-C. A Modular Chemoenzymatic Synthesis of Disialosyl Globopentaosylceramide (DSGb5Cer) Glycan. J. Org. Chem. 2020, 85, 15920–15935. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, Y.; Ito, A.; Kakoi, N.; Shimada, S.; Itoh, J.; Mitsuzuka, K.; Arai, Y. Ganglioside, disialosyl globopentaosylceramide (DSGb5), enhances the migration of renal cell carcinoma cells. Tohoku J. Exp. Med. 2015, 236, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Itoh, J.; Ito, A.; Shimada, S.; Kawasaki, Y.; Kakoi, N.; Saito, H.; Mitsuzuka, K.; Watanabe, M.; Satoh, M.; Saito, S. Clinicopathological significance of ganglioside DSGb5 expression in renal cell carcinoma. Glycoconj. J. 2017, 34, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, R.; Saito, S.; Bilim, V.; Hara, N.; Itoi, T.; Yamana, K.; Nishiyama, T.; Arai, Y.; Takahashi, K.; Tomita, Y. High incidence of GalNAc disialosyl lactotetraosylceramide in metastatic renal cell carcinoma. Anticancer. Res. 2007, 27, 4345–4350. [Google Scholar]
- Saito, S.; Orikasa, S.; Satoh, M.; Ohyama, C.; Ito, A.; Takahashi, T. Expression of globo-series gangliosides in human renal cell carcinoma. Jpn. J. Cancer Res. 1997, 88, 652–659. [Google Scholar] [CrossRef]
- Satoh, M.; Nejad, F.M.; Nakano, O.; Ito, A.; Kawamura, S.; Ohyama, C.; Saito, S.; Orikasa, S. Four new human renal cell carcinoma cell lines expressing globo-series gangliosides. Tohoku J. Exp. Med. 1999, 189, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Tringali, C.; Lupo, B.; Silvestri, I.; Papini, N.; Anastasia, L.; Tettamanti, G.; Venerando, B. The plasma membrane sialidase NEU3 regulates the malignancy of renal carcinoma cells by controlling β1 integrin internalization and recycling. J. Biol. Chem. 2012, 287, 42835–42845. [Google Scholar] [CrossRef]
- Saito, S.; Nojiri, H.; Satoh, M.; Ito, A.; Ohyama, C.; Orikasa, S. Inverse relationship of expression between GM3 and globo-series ganglioside in human renal cell carcinoma. Tohoku J. Exp. Med. 2000, 190, 271–278. [Google Scholar] [CrossRef][Green Version]
- Woutersen, R.A.; Appel, M.J.; van Garderen-Hoetmer, A.; Wijnands, M.V. Dietary fat and carcinogenesis. Mutat. Res. 1999, 443, 111–127. [Google Scholar] [CrossRef]
- Bojkova, B.; Winklewski, P.J.; Wszedybyl-Winklewska, M. Dietary Fat and Cancer-Which Is Good, Which Is Bad, and the Body of Evidence. Int. J. Mol. Sci. 2020, 21, 4114. [Google Scholar] [CrossRef]
- Gerber, M. Omega-3 fatty acids and cancers: A systematic update review of epidemiological studies. Br. J. Nutr. 2012, 107 (Suppl. S2), S228–S239. [Google Scholar] [CrossRef] [PubMed]
- Liotti, A.; Cosimato, V.; Mirra, P.; Cali, G.; Conza, D.; Secondo, A.; Luongo, G.; Terracciano, D.; Formisano, P.; Beguinot, F.; et al. Oleic acid promotes prostate cancer malignant phenotype via the G protein-coupled receptor FFA1/GPR40. J. Cell. Physiol. 2018, 233, 7367–7378. [Google Scholar] [CrossRef] [PubMed]
- Brasky, T.M.; Darke, A.K.; Song, X.; Tangen, C.M.; Goodman, P.J.; Thompson, I.M.; Meyskens, F.L., Jr.; Goodman, G.E.; Minasian, L.M.; Parnes, H.L.; et al. Plasma phospholipid fatty acids and prostate cancer risk in the SELECT trial. J. Natl. Cancer Inst. 2013, 105, 1132–1141. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Xiao, Y.; Yuan, Y.; Zhang, X.; Qin, C.; Xie, J.; Hao, Y.; Xu, T.; Wang, X. Effects of oleic acid on cell proliferation through an integrin-linked kinase signaling pathway in 786-O renal cell carcinoma cells. Oncol. Lett. 2013, 5, 1395–1399. [Google Scholar] [CrossRef]
- Xiang, F.; Wu, K.; Liu, Y.; Shi, L.; Wang, D.; Li, G.; Tao, K.; Wang, G. Omental adipocytes enhance the invasiveness of gastric cancer cells by oleic acid-induced activation of the PI3K-Akt signaling pathway. Int. J. Biochem. Cell Biol. 2017, 84, 14–21. [Google Scholar] [CrossRef]
- Soto-Guzman, A.; Navarro-Tito, N.; Castro-Sanchez, L.; Martinez-Orozco, R.; Salazar, E.P. Oleic acid promotes MMP-9 secretion and invasion in breast cancer cells. Clin. Exp. Metastasis 2010, 27, 505–515. [Google Scholar] [CrossRef]
- McCabe, A.J.; Wallace, J.M.; Gilmore, W.S.; McGlynn, H.; Strain, S.J. Docosahexaenoic acid reduces in vitro invasion of renal cell carcinoma by elevated levels of tissue inhibitor of metalloproteinase-1. J. Nutr. Biochem. 2005, 16, 17–22. [Google Scholar] [CrossRef]
- Bidoli, E.; Talamini, R.; Zucchetto, A.; Polesel, J.; Bosetti, C.; Negri, E.; Maruzzi, D.; Montella, M.; Franceschi, S.; La Vecchia, C. Macronutrients, fatty acids, cholesterol and renal cell cancer risk. Int. J. Cancer 2008, 122, 2586–2589. [Google Scholar] [CrossRef] [PubMed]
- Brock, K.E.; Gridley, G.; Chiu, B.C.; Ershow, A.G.; Lynch, C.F.; Cantor, K.P. Dietary fat and risk of renal cell carcinoma in the USA: A case-control study. Br. J. Nutr. 2009, 101, 1228–1238. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.E.; Spiegelman, D.; Hunter, D.J.; Albanes, D.; Bernstein, L.; van den Brandt, P.A.; Buring, J.E.; Cho, E.; English, D.R.; Freudenheim, J.L.; et al. Fat, protein, and meat consumption and renal cell cancer risk: A pooled analysis of 13 prospective studies. J. Natl. Cancer Inst. 2008, 100, 1695–1706. [Google Scholar] [CrossRef]
- Schwenk, R.W.; Holloway, G.P.; Luiken, J.J.; Bonen, A.; Glatz, J.F. Fatty acid transport across the cell membrane: Regulation by fatty acid transporters. Prostaglandins Leukot Essent Fat. Acids 2010, 82, 149–154. [Google Scholar] [CrossRef] [PubMed]
- Schaffer, J.E.; Lodish, H.F. Expression cloning and characterization of a novel adipocyte long chain fatty acid transport protein. Cell 1994, 79, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Hara, T.; Kimura, I.; Inoue, D.; Ichimura, A.; Hirasawa, A. Free fatty acid receptors and their role in regulation of energy metabolism. Rev. Physiol. Biochem. Pharmacol. 2013, 164, 77–116. [Google Scholar]
- Hopkins, M.M.; Meier, K.E. Free Fatty Acid Receptors and Cancer: From Nutrition to Pharmacology. Handb. Exp. Pharmacol. 2017, 236, 233–251. [Google Scholar] [PubMed]
- Hopkins, M.M.; Meier, K.E. Free fatty acid receptor (FFAR) agonists inhibit proliferation of human ovarian cancer cells. Prostaglandins Leukot Essent Fat. Acids 2017, 122, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, M.M.; Zhang, Z.; Liu, Z.; Meier, K.E. Eicosopentaneoic Acid and Other Free Fatty Acid Receptor Agonists Inhibit Lysophosphatidic Acid- and Epidermal Growth Factor-Induced Proliferation of Human Breast Cancer Cells. J. Clin. Med. 2016, 5, 16. [Google Scholar] [CrossRef]
- Wang, J.; Hong, Y.; Shao, S.; Zhang, K.; Hong, W. FFAR1-and FFAR4-dependent activation of Hippo pathway mediates DHA-induced apoptosis of androgen-independent prostate cancer cells. Biochem. Biophys. Res. Commun. 2018, 506, 590–596. [Google Scholar] [CrossRef]
- Takahashi, K.; Fukushima, K.; Onishi, Y.; Minami, K.; Otagaki, S.; Ishimoto, K.; Fukushima, N.; Honoki, K.; Tsujiuchi, T. Involvement of FFA1 and FFA4 in the regulation of cellular functions during tumor progression in colon cancer cells. Exp. Cell Res. 2018, 369, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Monaco, M.E. Fatty acid metabolism in breast cancer subtypes. Oncotarget 2017, 8, 29487–29500. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tan, S.K.; Hougen, H.Y.; Merchan, J.R.; Gonzalgo, M.L.; Welford, S.M. Fatty acid metabolism reprogramming in ccRCC: Mechanisms and potential targets. Nat. Rev. Urol. 2022, 20, 48–60. [Google Scholar] [CrossRef]
- Alo, P.L.; Visca, P.; Marci, A.; Mangoni, A.; Botti, C.; Di Tondo, U. Expression of fatty acid synthase (FAS) as a predictor of recurrence in stage I breast carcinoma patients. Cancer 1996, 77, 474–482. [Google Scholar] [CrossRef]
- Epstein, J.I.; Carmichael, M.; Partin, A.W. OA-519 (fatty acid synthase) as an independent predictor of pathologic state in adenocarcinoma of the prostate. Urology 1995, 45, 81–86. [Google Scholar] [CrossRef]
- Rashid, A.; Pizer, E.S.; Moga, M.; Milgraum, L.Z.; Zahurak, M.; Pasternack, G.R.; Kuhajda, F.P.; Hamilton, S.R. Elevated expression of fatty acid synthase and fatty acid synthetic activity in colorectal neoplasia. Am. J. Pathol. 1997, 150, 201–208. [Google Scholar] [PubMed]
- Nguyen, P.L.; Ma, J.; Chavarro, J.E.; Freedman, M.L.; Lis, R.; Fedele, G.; Fiore, C.; Qiu, W.; Fiorentino, M.; Finn, S.; et al. Fatty acid synthase polymorphisms, tumor expression, body mass index, prostate cancer risk, and survival. J. Clin. Oncol. 2010, 28, 3958–3964. [Google Scholar] [CrossRef] [PubMed]
- Horiguchi, A.; Asano, T.; Asano, T.; Ito, K.; Sumitomo, M.; Hayakawa, M. Pharmacological inhibitor of fatty acid synthase suppresses growth and invasiveness of renal cancer cells. J. Urol. 2008, 180, 729–736. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Hu, X.; Anwaier, A.; Wang, J.; Liu, W.; Tian, X.; Zhu, W.; Ma, C.; Wan, F.; Shi, G.; et al. Fatty Acid Synthase Correlates With Prognosis-Related Abdominal Adipose Distribution and Metabolic Disorders of Clear Cell Renal Cell Carcinoma. Front. Mol. Biosci. 2020, 7, 610229. [Google Scholar] [CrossRef]
- Yuan, Y.; Yang, X.; Li, Y.; Liu, Q.; Wu, F.; Qu, H.; Gao, H.; Ge, J.; Xu, Y.; Wang, H.; et al. Expression and prognostic significance of fatty acid synthase in clear cell renal cell carcinoma. Pathol. Res. Pract. 2020, 216, 153227. [Google Scholar] [CrossRef]
- Che, L.; Paliogiannis, P.; Cigliano, A.; Pilo, M.G.; Chen, X.; Calvisi, D.F. Pathogenetic, Prognostic, and Therapeutic Role of Fatty Acid Synthase in Human Hepatocellular Carcinoma. Front. Oncol. 2019, 9, 1412. [Google Scholar] [CrossRef]
- Lu, T.; Sun, L.; Wang, Z.; Zhang, Y.; He, Z.; Xu, C. Fatty acid synthase enhances colorectal cancer cell proliferation and metastasis via regulating AMPK/mTOR pathway. Onco Targets Ther. 2019, 12, 3339–3347. [Google Scholar] [CrossRef][Green Version]
- Chang, L.; Fang, S.; Chen, Y.; Yang, Z.; Yuan, Y.; Zhang, J.; Ye, L.; Gu, W. Inhibition of FASN suppresses the malignant biological behavior of non-small cell lung cancer cells via deregulating glucose metabolism and AKT/ERK pathway. Lipids Health Dis. 2019, 18, 118. [Google Scholar] [CrossRef]
- Wettersten, H.I.; Hakimi, A.A.; Morin, D.; Bianchi, C.; Johnstone, M.E.; Donohoe, D.R.; Trott, J.F.; Aboud, O.A.; Stirdivant, S.; Neri, B.; et al. Grade-Dependent Metabolic Reprogramming in Kidney Cancer Revealed by Combined Proteomics and Metabolomics Analysis. Cancer Res. 2015, 75, 2541–2552. [Google Scholar] [CrossRef]
- Albiges, L.; Hakimi, A.A.; Xie, W.; McKay, R.R.; Simantov, R.; Lin, X.; Lee, J.L.; Rini, B.I.; Srinivas, S.; Bjarnason, G.A.; et al. Body Mass Index and Metastatic Renal Cell Carcinoma: Clinical and Biological Correlations. J. Clin. Oncol. 2016, 34, 3655–3663. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Z.; Huang, C.; Sun, J.; Qiu, W.; Zhang, J.; Li, H.; Jiang, T.; Huang, K.; Cao, J. RNA interference-mediated signal transducers and activators of transcription 3 gene silencing inhibits invasion and metastasis of human pancreatic cancer cells. Cancer Sci. 2007, 98, 1099–1106. [Google Scholar] [CrossRef] [PubMed]
- Slade, R.F.; Hunt, D.A.; Pochet, M.M.; Venema, V.J.; Hennigar, R.A. Characterization and inhibition of fatty acid synthase in pediatric tumor cell lines. Anticancer. Res. 2003, 23, 1235–1243. [Google Scholar] [PubMed]
- Wang, X.; Du, G.; Wu, Y.; Zhang, Y.; Guo, F.; Liu, W.; Wu, R. Association between different levels of lipid metabolismrelated enzymes and fatty acid synthase in Wilms’ tumor. Int. J. Oncol. 2020, 56, 568–580. [Google Scholar]
- Camassei, F.D.; Jenkner, A.; Rava, L.; Bosman, C.; Francalanci, P.; Donfrancesco, A.; Alo, P.L.; Boldrini, R. Expression of the lipogenic enzyme fatty acid synthase (FAS) as a predictor of poor outcome in nephroblastoma: An interinstitutional study. Med. Pediatr. Oncol. 2003, 40, 302–308. [Google Scholar] [CrossRef]
- Quan, J.; Bode, A.M.; Luo, X. ACSL family: The regulatory mechanisms and therapeutic implications in cancer. Eur. J. Pharmacol. 2021, 909, 174397. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research, N. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 2013, 499, 43–49. [Google Scholar] [CrossRef]
- Zhang, C.Y.; Hu, H.L.; Huang, R.Z.; Huang, G.M.; Xi, X.Q. ACSL3 is a potential prognostic biomarker for immune infiltration in clear cell renal cell carcinoma. Front. Surg. 2022, 9, 909854. [Google Scholar] [CrossRef]
- Klasson, T.D.; LaGory, E.L.; Zhao, H.; Huynh, S.K.; Papandreou, I.; Moon, E.J.; Giaccia, A.J. ACSL3 regulates lipid droplet biogenesis and ferroptosis sensitivity in clear cell renal cell carcinoma. Cancer Metab. 2022, 10, 14. [Google Scholar] [CrossRef]