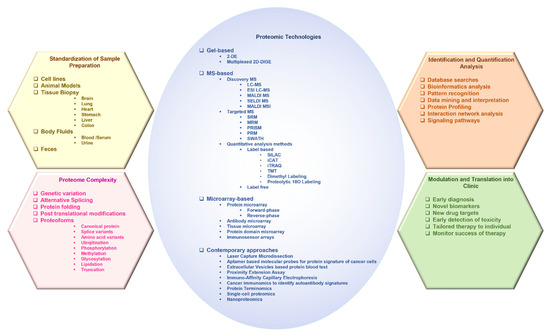
2. Advances in Proteomic Technologies Used in the Study of Cancer
2.1. Gel-Based Approaches
2.1.1. Two-Dimensional Gel Electrophoresis
Two-dimensional gel electrophoresis (2-DE) is an important and well-established technical platform for the reliable and efficient separation of proteins based on relative mass (Mr) and charge
[30][38]. The conventional concepts of 2-DE combine isoelectric focusing (pI) with sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), which results in the resolution of thousands of spots in one gel. High-resolution 2-DE can even resolve up to 10,000 protein spots (including separation of proteoforms) per gel
[17][31][22,39].
2.1.2. 2D Differential in-Gel Electrophoresis
The 2D differential in-gel electrophoresis (2D DIGE) allows parallel comparison of multiple protein samples within the same gel, thus facilitating relative comparison of different sample states without gel-to-gel variability. For quantitative analysis by DIGE, the protein samples are labeled with spectrally distinct, charge- and mass-matched fluorescence dyes, such as Cy2, Cy3, or Cy5, and mixed before electrophoresis and run along with a differently labeled standard on the same 2D gel. The individual protein products are differentially visualized by differential fluorescence
[32][33][44,45]. For identification, the gel-separated protein products can be either probed with antibodies, or digested into peptides to obtain a peptide mass fingerprint that can be examined against theoretical fingerprints of protein sequences in the database, or subjected to high-resolution mass spectrometry (MS) for accurate mass determination
[34][35][36]. Moreover, 2-DE has been used in proteome analysis of human tissue, plasma, and serum, with or without prior fractionation [36][37]. DIGE has improved accuracy over 2-DE and can be utilized in biomarker discovery that does not involve high-throughput sample processing [38][39][40][41].[46,47,48]. Moreover, 2-DE has been used in proteome analysis of human tissue, plasma, and serum, with or without prior fractionation [48,50]. DIGE has improved accuracy over 2-DE and can be utilized in biomarker discovery that does not involve high-throughput sample processing [49,51,52,53].
2.2. Mass Spectrometry-Based Approaches
Mass spectrometry (MS) is an analytical tool that is used to measure the mass-to-charge ratio (
m/z) of one or more molecules present in a sample. The results are obtained as a mass spectrum, which is a plot of ion signal (the intensity) as a function of the
m/z ratio. These spectra are used to determine the elemental or isotopic signature, exact molecular masses of the sample components, and elucidate the chemical identity or structure of molecules. Thus, MS can be used to (1) identify unknown compounds by determining molecular weight, (2) quantify known compounds, and (3) elucidate the structure and chemical properties of molecules
[42][43][54,55]. Moreover, it can be applied to pure samples as well as complex mixtures.
A mass spectrometer typically consists of three major components: an ion source, a mass analyzer, and a detector. In a typical MS procedure, the sample whether solid, liquid, or gaseous is first ionized, and magnetic and/or electric fields are used to separate ions by virtue of their different trajectories (based on their
m/z ratio) in a vacuum that is finally detected by the detector. Ion source: Each phase (solid, liquid, or gaseous) requires different ionization methods. The ionization may be continuous or pulsed and may occur at different pressures. The ions generated may be positively or negatively charged. In biomedical applications, samples are predominantly liquids containing large molecules that require continuous soft ionization to avoid fragmentation, such as electrospray ionization (ESI) or matrix-assisted laser desorption/ionization (MALDI). Mass analyzer: The mass analyzers can use magnetic and/or electric fields with a static or time-varying field, and operation is made continuous or cyclic. The main variants of mass analyzers include the magnetic sector, Fourier-transform ion cyclotron resonance, quadrupole, ion trap, and time-of-flight (TOF) mass spectrometer. Usually, electric fields are preferred because they avoid the requirement of a large, heavy magnet. Subsequently, the quadrupole, ion trap and TOF mass spectrometer are preferred
[44][56]. All offer high performance with several advantages, such as sensitivity, mass resolution, and mass range based on the requirement. Sensitivity limits are set by the ion flux and space charge effects at low and high fluxes; the mass resolution is limited by the thermal spread in ion velocity and the precision of the applied fields, while the mass range is limited by the magnitude of the field. Detector: The final element of the MS is a detector that records the charge induced or the current produced when an ion passes by or hits a surface.
2.2.1. Liquid Chromatography–Mass Spectrometry
In liquid chromatography–mass spectrometry (LC-MS) systems, the liquid analytes are first separated by LC and individual molecules are passed sequentially into the mass spectrometer to identify their masses. LC variants with an increase in pressure include high-performance LC (HPLC) and ultra-performance LC (UPLC). The LC column effluent is nebulized, desolvated, and softly ionized using ESI, creating charged particles. In ESI, the solubilized sample is passed through a high-voltage needle held at atmospheric pressure that produces charged droplets, which destabilize and explode into finer droplets. The desolvated analyte ions migrate under a high vacuum through a mass analyzer that separates ions based on their
m/z ratio and transfers them into a detector. LC-MS instrument is usually an HPLC unit with an attached mass spectrometer and LC-MS/MS is an HPLC with two mass spectrometers.
Tandem MS (
MS/
MS) consists of two mass analyzers that have been shown to improve speed and sensitivity and are used for the analysis of protein or peptide mixtures or the determination of the mass of intact protein product. It is commonly used for proteome analysis of complex biological samples (such as human serum or feces) where the overlap between peptide masses cannot be resolved with a high-resolution mass spectrometer. The first mass analyzer is used to isolate the precursor ions that are subsequently fragmented in a collision cell. The resulting fragment ions are then separated in the second mass spectrometer, generating a pattern of fragments (the tandem mass spectrum), which forms the characteristic fingerprint of the molecule of interest. The most popular mass analyzers used in tandem MS include quadrupole (Q), time-of-flight (TOF), or hybrid analyzers, such as quadrupole coupled with TOF (Q-TOF), depending on the data required (structural or quantitative), resolution, and mass accuracy.
The quadrupole mass analyzer consists of four parallel cylindrical metal rods at a well-defined distance from each other. A combination of direct current (DC) and radio frequency (RF) voltages is then applied to the rods, creating a time-varying quadrupolar field that separates ions based on the stability of their trajectories. At a particular ratio of DC to RF voltage, ions with specific
m/z will have confined trajectories and without discharging will pass through the length of the quadrupole. The disadvantage of quadrupole is that length, constructional precision, the frequency of the RF voltage limits its mass selectivity, and the amplitude of the RF voltage limits mass range
[44][45][56,60].
The TOF mass analyzer provides high mass accuracy and range. In the ion modulator region of the TOF analyzer, ions are accelerated under an electric field to acquire similar kinetic energy and then admitted to a field-free drift region of the flight tube for mass separation. Ions become separated based on their
m/z value by measuring the time taken to traverse a known distance before striking a detector. The lighter ions travel faster and the heavier ions take longer to travel, as the square of the drift time of an ion is proportional to its
m/
z ratio. A mass spectrum is generated, representing the number of ions hitting the detector over time. A full mass spectrum can be obtained by scans of the whole mass range, which enables the determination of the molecular masses of the ions with high accuracy. High mass range/resolution can be obtained by short pulse, low axial velocity, and large distance (the length of the flight path). However thermal energy causes uncertainty in the initial position and velocity of the ions which can be optimized by orthogonal acceleration, delayed ion extraction, or using a reflectron (reflection by a stacked electrode) to reach a much higher resolution than linear TOF.
Q-TOF MS is a hybrid mass analyzer that advantageously combines the ion selection properties of a quadrupole with the high speed, mass resolution, and accuracy of a TOF in a single system. It usually has two quadrupole systems and a TOF tube. The first quadrupole acts as a mass filter for the selection of specific ions based on their
m/z ratio and the second quadrupole acts as a collision cell where ions are bombarded by inert gas molecules, such as nitrogen or argon, resulting in the fragmentation of the ions by a process known as collision-induced dissociation (CID). In wide band pass/RF only mode, there is no gas in the collision cell and all ions from the quadrupole are transferred into the TOF analyzer without subsequent fragmentation of ions. However, in narrow pass mode, fragmentation of a selected ion with a known
m/z value occurs and the quadrupole acts as a filter to pass ions with a particular
m/z value into the TOF analyzer. Most ions produce a signature fragmentation pattern that can be identified using databases or chemical standards. The ions with the same mass can be differentiated based on their fragmentation pattern. The Q-TOF offers high mass accuracy together with tandem MS, which is suitable for nontargeted profiling applications
[46][47][64,65].
2.2.2. Matrix-Assisted Laser Desorption/Ionization
Matrix-assisted laser desorption/ionization (MALDI) is a soft ionization technique used in MS that involves a laser collision with a matrix of molecules to make the analyte molecules into the gas phase without fragmenting or decomposing them. It is suitable for identification and spatial distribution studies of large biomolecules, which are either non-volatile or thermally unstable. The analyte dissolved in a solvent containing a selected matrix, such as sinapic acid or α-cyano-4-hydroxycinnamic acid, is deposited on a target plate for drying and crystallization. In a variant of MALDI called SALDI (surface-assisted laser desorption/ionization), the solid nanomaterial is used as the matrix, which provides a more homogeneous sample distribution. The target plate is then placed in the vacuum chamber of a mass spectrometer and bombarded by photons from a pulsed laser, resulting in the desorption and ionization of the matrix. The energy from the matrix is gently transferred to the sample molecules leaving it intact and in the gas phase, yielding protonated (cationized) or deprotonated (anionized) molecular ions. The ions are then separated based on their TOF which is proportional to their m/z value. MALDI-TOF MS a method of choice in clinical settings for the identification of biomolecules in complexes [48][49][66,67] and cancer diagnosis and prognosis [50][68].
2.2.3. MALDI Mass Spectrometry Imaging
MALDI mass spectrometry imaging (MALDI-MSI) is a powerful technique by which the spatial and temporal distribution of proteoforms and biomolecules can be investigated directly from a tissue section without the need for extraction, purification, and separation measures
[51][79]. The MSI is based on the mapping of the corresponding ion intensities along with the determination of the spatial distribution of many molecules in a sample. In MALDI-MSI, a tissue section is coated with matrix and the sample is raster-scanned (with a spatial resolution ranging from approximately 200 μm down to 20 μm) in the mass spectrometer resulting in spatially resolved mass spectra. The laser only strikes the matrix crystals without affecting the tissue section. After the MALDI measurement, histological staining allows a histology-directed analysis of the mass spectra. To reduce analyte diffusion which alters the original distribution and reduces the spatial resolution, matrix-free ionization platforms have been developed for use, such as inorganic matrix and nanophotonic platforms instead of organic matrices
[52][80]. MALDI-MSI is been extensively employed in clinical proteomics in cancer
[53][54][55][91,92,93].
2.2.4. Surface-Enhanced Laser Desorption/Ionization Time-Of-Flight Mass Spectrometry
The Surface-enhanced laser desorption/ionization time-of-flight mass spectrometry (SELDI-TOF-MS) technique is one variant of MALDI that uses surface extraction by ProteinChip. It was introduced in 1993
[56][99] and later commercialized as the ProteinChip system by Ciphergen Biosystems in 1997. In SELDI, the sample containing the protein mixture is applied on a surface customized with a chemical functionality, such as binding affinity. The different substances used to modify SELDI surface may include antibodies, receptors, ligands, nucleic acids, carbohydrates, metal ions, or chromatographic surfaces (e.g., cationic, anionic, hydrophobic, or hydrophilic exchangers). The protein product of interest becomes sequestered by interacting with substances on the surface of ProteinChip based on biological or chemical affinities. The nonspecific substances and contaminants are removed by subsequent on-spot washing and only the surface-bound protein products are left for analysis. An energy absorbing matrix (e.g., SPA) is applied to the surface for crystallization with the target molecules and then subjected to laser ionization, which delivers higher specificity and sensitivity in subsequent analysis. SELDI-TOF MS has been employed for the diagnosis, detection, and identification of biomarker candidates for various cancer types
[57][58][59][60][61][62][63][64][65][101,102,103,104,105,106,107,110,111].
2.2.5. Targeted/Directed Mass Spectrometry
The characteristic of targeted MS is the selection and fragmentation of a predetermined set of precursor ions that are either predicted or identified in a survey scan. The multiple stages of tandem MS are utilized for two or three ions of a specific mass at a specific time. The
m/z values and time can be defined in an inclusion list that is derived from a previous analysis. The specific targeting of analyte peptides of interest provides exquisite specificity and sensitivity. In contrast to discovery MS, protein identification based on fragment ion spectra and protein quantification based on survey scans is decoupled and performed in two separate experiments
[66][67][68][116,117,118]. Targeted MS is manifold sensitive than discovery MS and provides highly precise quantification using internal standards [
118]. It is a widely adopted approach in oncoproteomics analysis
[69][70][71][72][73][74][126,130,132,145,146,147].
2.2.6. Quantitative Analysis Methods
The most attractive part of proteomics is its ability to reveal novel biomarkers of cancer. With the progression of cancer, changes in proteoforms and their differential distribution both in tissues and body fluids can be monitored via concurrent qualitative and quantitative profiling of numerous proteoforms. Accurate quantitation is crucial for oncoproteomics analysis. For quantitative investigations in clinical research, label-based and label-free approaches are widely used. In the case of label-based approaches, isotopic labeling is used, which involves in vivo or in vitro incorporation of stable isotopes into proteins or peptides for comparative analysis with isotope-free markers. Labeling allows multiplexing that permits simultaneous analysis of several samples and reduces experimental variability inherent in sample processing.
2.3. Microarrays
Microarrays, also known as biochips, are a collection of microscopic biomolecules spotted on a solid support that are used to identify interacting partners via affinity interaction.
2.3.1. Protein Microarray
A protein microarray (or protein chip) is a high-throughput tool for studying the biochemical activities of proteins, their interactions, and function determination on a large scale using miniaturized assays
[75][260]. The main advantage is that large numbers of proteins can be followed in parallel. Typically, the chip contains numerous spots of either proteins or their ligands arranged in a predefined pattern, arrayed by robots onto a solid support surface. The support surface can be a glass slide, nitrocellulose membrane, bead, or microtiter plate, to which an array of capture proteins is bound
[76][261]. Usually, fluorescent dye-labeled probe molecules are added to the array after sample application. Any reaction between the probe and the immobilized proteins emits a fluorescent signal that is measured by a laser scanner. Protein microarrays are quick, automated, cost-effective (require minuscule samples and reagents), and highly sensitive. Protein microarrays have become an indispensable tool for proteomic applications and multi-parameter clinical diagnostic tests
[77][262].
2.3.2. Antibody/Antigen Microarrays
In antibody microarrays, the specific capture antibodies are immobilized on a modified planar solid surface such as a nitrocellulose membrane, glass slide, silicone, or bead via covalent binding, affinity binding, or physical entrapment. The sample (such as serum or cell lysate) is then applied to detect the interaction between the antibody and its target protein. Antibody arrays, such as bead-based arrays and sandwich ELISA-based planar arrays, provide medium-/low-plex proteomic profiling. For high-plex profiling, samples are labeled with fluorescent, chemiluminescent, or oligo-coupled tags to allow differential signal amplification and detection. This method can practically characterize over a thousand proteins with minimal immunogenic cross-reactivity induced by antibodies
[78][59].
Antibody arrays have very high performance for knowledge-based examinations, providing a high-throughput, semi-quantitative, or quantitative analysis. In contrast to untargeted proteomic approaches, it is highly sensitive. Ultramicroarrays have been developed to combine the advantages of multiplexing capabilities, higher throughput, and cost savings, with the ability to screen minuscule samples
[79][272]. Antibody arrays are particularly useful for proteomic profiling of low-abundance proteoforms. It has been extensively applied in the high-throughput multiplexed analysis of cancer biomarkers
[80][81][273,274].
2.3.3. Tissue Microarrays
The tissue microarray (TMA) is a high-throughput technology that enables simultaneous proteome analysis from thousands of individual tissue samples in a single microscopic slide
[82][283]. It was first described by Kononen in 1998
[83][284]. The tissues are formalin-fixed and paraffin-embedded from which small cylindrical tissue cores as small as 0.6 mm in diameter from regions of interest that are extracted using hollow needles of set diameters and transferred into a matrix slot within a recipient paraffin block. Sections from each microarray array block are cut using a microtome into 50–1000 sections that can be subjected to independent tests on a microscope slide and analyzed by a variety of assay and staining techniques, including immunohistochemistry (IHC) and fluorescent in situ hybridization (FISH) analysis, in situ PCR, and cDNA hybridization. This facilitates the rapid analysis of hundreds of patient samples
[84][285].
2.3.4. Protein Domain Microarray
Protein microarrays are efficient in high-throughput identification and quantification of protein–protein interactions. However, proteins exhibit a wide range of physicochemical properties and often recombinant production is difficult. To sidestep these issues and to read the PTM signal placed on the interacting partners, families of protein interaction domains can be focused. Protein domains bind to short peptide motifs in their corresponding ligands to mediate protein–protein interactions. These peptide recognition elements are important for multiprotein complex assemblies. The protein domain microarray consists of protein interaction domains arrayed onto solid support, such as nitrocellulose-coated glass slides, to generate a protein–domain chip
[85][292]. The arrayed domains retain their binding integrity for their respective peptides/protein. The high-throughput quantification of domain–peptide interactions can be performed using fluorescently labeled synthetic peptides
[86][293].
2.3.5. Immunosensor Arrays
Immunosensor arrays are a type of affinity-based biosensors that detect a specific target analyte or antigen by the formation of a stable immunocomplex between the antigen and the capture antibody, which results in the generation of a measurable signal by a transducer. The use of antibodies as molecular recognition agents provides ultrahigh specificity in immunoassay and facilitates the detection of cancer biomarkers
[87][295]. For cancer diagnostics, the immunoassay is integrated with several detection strategies, such as fluorescence
[88][296], colorimetric
[89][297], plasmon resonance sensors
[90][298], electrical
[91][299], optical
[91][299], electrochemical
[92][300], chemiluminescence
[93][301], and electrochemiluminescence
[94][302].
One aspect of oncoproteomics is directed toward the development of accessible and ultra-sensitive cancer diagnostic tools that rely on protein biomarkers associated with various cancer that are overexpressed in body fluids. Protein biomarker detection for point-of-care use requires highly sensitive, non-invasive microfluidic cancer diagnostics that can overcome the limitation of low sensitivities imposed by imaging and invasive biopsies. Electrochemical immunoassays have become popular as protein detection methods due to their inherent high sensitivity and ease of coupling with 3D printed electrodes. Integrated chips with printed electrodes can be built at a low cost and designed for automation. Three-dimensional printing also known as additive manufacturing is being utilized to develop user-friendly, semi-automated, and highly sensitive protein biomarker sensors at low-cost. These can be tailored toward clinical needs
[95][303]. Most of these ultrasensitive detection systems use enzyme-linked immunosorbent assay (ELISA) features with microfluidics that permits easy manipulation and good fluid dynamics to deliver reagents and detect the desired proteins
[96][304].
3. Contemporary Technologies and Approaches
3.1. Laser Capture Microdissection
Laser capture microdissection (LCM) is an effective extraction technique to harvest pure subpopulations of cells from tissue sections under direct visualization of a microscope. The cells of interest are harvested either directly or by cutting away unwanted cells to obtain histologically pure enriched specific cell populations. LCM has expanded the analytical capabilities of proteomics to analyze proteins from extremely small samples
[97][98][309,310]. It basically allows for the miniaturization of extraction, isolation, and detection of hundreds of proteins from different cell populations containing only a few cells. However, as the sample size decreases, each step requires care. The LCM dissected tissues are subjected to protein extraction, reduction, alkylation, and digestion, followed by injection into a nano-LC MS/MS system to simultaneously identify and quantify hundreds of proteins. The validation can be performed by secondary screening using immunological techniques, such as IHC or immunoblots
[97][99][309,311]. The advancement in LCM technology enables effective high-throughput sampling of specific cellular subtypes
[100][311]. LCM-coupled 2D-DIGE and/or quantitative MS approaches have been used for proteomics analysis of distinct, pure cell populations
[101][102][103][312,313,314] and to investigate various cancer-associated protein profiles
[99][104][103][105][106][311,313,314,315,316].
3.2. Aptamer-Based Molecular Probes for Protein Signature of Cancer Cells
Aptamers are a class of short, single-stranded DNA, RNA, or peptide (~25–80 nucleotides/amino acids) that after acquiring a specific tertiary structure, bind to various targets with high affinity and selectivity. Aptamers are also known as a ‘chemical antibody’ and possess several intrinsic advantages, such as convenient modification, easy synthesis, good compatibility, and high programmability. A process known as systematic evolution of ligands by exponential enrichment (SELEX) was first used to screen aptamers. Aptamers can be generated against various targets, such as small molecules, peptides, proteins, and intact living cancer cells. Some of the examples of aptamers used against cancer cells are Sgc8, specific for acute lymphoblastic leukemia
[107][317] and XQ-2D, for pancreatic ductal adenocarcinoma
[108][318].
3.3. Extracellular Vesicle-Based Protein Blood Test
Tumor-derived Extracellular Vesicles (EVs) have recently emerged as an important biomarker in blood circulation for the diagnosis of cancer. The EVs are nano-/micro-meter size lipid bilayer-enclosed vesicles that contain various molecules, such as proteins, nucleic acid, and lipids from parental tumor cells. Tumor-derived EVs are present in abundance in blood circulation compared to other biomarkers, as they release in blood 10
4 quantities per day. The proteins enveloped in EVs play an important role in cancer metastasis and progression, including immune evasion, matrix remodeling, tumor vascularization, and premetastatic niche formation. Detection of EV protein markers facilitate the diagnosis and monitoring of various cancers
[109][110][111][322,323,324].
3.4. Proximity Extension Assay
The proximity extension assay (PEA) is a combination of two sandwich ELISAs and highly specific and sensitive polymerase chain reaction (PCR) technologies that detect protein–protein interaction and liquid biopsy-based discovery in cancer. It has a broad dynamic range of 10 logs and minimal sample requirement, which makes it a very useful tool for serological profiling. In PEA, multiple antibodies are pooled with the protein of interest. Each antibody in a pair is attached with complementary DNA oligonucleotides that allow hybridization when the correct antibody pairs come close together by binding to the target protein. The resultant double-stranded DNA is PCR amplified and is used to measure the relative concentration of the target proteins. The most recent commercial PEA assay has standard measurement coverage of 3072 target proteins. PEA is being increasingly used to identify biomarkers in certain cancers
[112][113][329,331].
3.5. Immuno-Affinity Capillary Electrophoresis
Immuno-affinity capillary electrophoresis (IACE) is an emerging powerful diagnostic tool to isolate, separate, detect, and characterize proteoform in biological fluids. It combines the power of highly selective capture agents with the high resolving power of capillary electrophoresis. IACE separates the substances by specific and non-specific noncovalent affinity interactions during electrophoresis. In one of the variant of IACE, the interacting target molecule is captured and bound to affinity reagents onto the wall of capillary or solid support. Then, the remaining sample is removed, and the target molecule is released using an elution buffer [
337]. It is used as a point-of-care instrument to profile proteoform patterns in biological fluids and tumor cells
[114][115][116][117][118][119][336,337,338,339,340,341].
3.6. Cancer Immunomics to Identify Autoantibody Signatures
Antibodies associated with cancer develop early during carcinogenesis when cancer-associated antigens appear in premalignant or malignant tissue. The cancer antigens are recognized by the effective immune response of autoantibodies, which makes autoantibodies a suitable biomarker for cancer detection. For example, autoantibodies against HCC1, CDKN2A, P53, the cellular inhibitor of PP2A (CIP2A), and the cyclin-dependent kinase inhibitor 2A (CDKN2A) indicate the presence of HCC prior to its clinical diagnosis
[120][342]. These autoantibodies can be detected by proteome analysis in serum by using 2DGE, immunoblotting, image analysis, and MS
[120][342].
3.7. Protein Terminomics
Proteases are key enzymes involved in protein terminomics. Proteases regulate vital biological processes of apoptosis, neurodegeneration, infection, and cell differentiation. Proteolysis performed by proteases is an important post-translational modification of a protein. Around 600 human proteases are reported and categorized into five families based on their catalytic mechanisms (threonine, serine, cysteine, metallo, and aspartyl proteases). There are two methods commonly used in protein terminomics: N-terminomics and C-terminomics. N-terminomics involves the labeling of the proteolytic protein fragments and the enrichment of the fragments from the complex mixture. The enrichment can be achieved by adding the functional group or by labeling with the isotope to the cleaved peptide. As C-terminal labeling is quite difficult as compared with N-terminal labeling, the N-terminomics is widely used. In N-terminomics, well established methods include the COFRADIC (combinatorial fractional diagonal chromatography), subtilligase, and TAILS (terminal amine isotopic labeling of substrates) methods
[121][344]. N-terminomics has been used to identify the substrate of neutrophil-specific membrane-type 6 matrix metalloproteinase (MTP6-MMP), which plays a role in cancer
[122][345]. Moreover, Alcaraz et al. used TAILS to identify the substrate of cathepsin D, which is a tumor-specific protease in triple-negative breast cancer cells
[123][346].
3.8. Single-Cell Proteomics
Cancer tissue shows multiple genomic variations and heterogeneity at the level of proteome. This cell-to-cell variability is responsible for the altered biomarker expression in the different cells of the same tissue that may be overlooked when biomarker quantitation is based on the bulk tissue sample. Single-cell proteomics allows measuring of the level of prognostic and diagnostic biomarkers at the level of a single cell of a cancerous tissue that provides information about a single-cell subpopulation carrying cancerous characteristics
[124][125][347,348]. This information can further be used for patient risk stratification and individualized therapy. The techniques which are being used for single-cell proteomics are as follows: microfluidics and laboratory-on-a-chip technology, flow cytometry, mass cytometry, and chemical cytometry.
3.9. Nanoproteomics
To detect the low abundance proteoforms that can be isolated from the limited source material (e.g., biopsies), the nanoproteomics platform provides improved specificity, reproducibility, biocompatibility, and robustness compared to the current conventional proteomic techniques. Nanoproteomics can be defined as the application of nanobiotechnology to proteomics. It uses nanoscale devices such as nanofluidics and nanoarrays. Unique nanomaterials such as quantum dots (QDs), carbon nanotubes (CNTs), and gold nanoparticles (GNPs) are being used in nanoproteomics techniques. The application of nanoproteomics techniques in cancer advances the discovery of biomarkers and detection of early cancer pathogenesis
[126][349].
3.10. PTM Enrichment Methods
Post-translation modifications (PTMs) comprise phosphorylation, acetylation, methylation, glycosylation, ubiquitination, and SUMOylation (among other modifications), and because of their low abundance and labile nature, enrichment of a modified protein product is required for MS analysis.
The enrichment of proteoforms can be achieved by affinity or chemical strategies prior to MS. Affinity strategies require antibody/protein domain recognition for purification or chromatographic separation based on specific properties of the PTM, while chemical enrichment strategies involve chemo-selective probes, metabolic labeling by unnatural precursors, and chemoenzymatic labeling. For instance, phosphopeptide enrichment by affinity approaches depends on the interaction of phosphorylated amino acid with different binding reagents and is categorized into ion exchange chromatography, affinity chromatography, and antibody/protein domain-based enrichment of phosphor-tyrosines
[127][128][354,355].
4. Role of Proteomics in the Prognosis and Diagnosis of Cancer
Proteomics investigations can be divided into two major areas: expression proteomics and functional proteomics. Expression proteomics deals with the up and downregulation of protein levels. Functional proteomics defines the molecular mechanism and discovers the unknown function of a protein
[129][130][356,357]. It includes PTMs, characterization of protein complexes, and enzyme activities. The detection of various proteoforms by functional proteomics helps to identify therapeutic targets and diagnostic biomarkers in cancer.
HCC is diagnosed by liver biopsy analysis or by cross-sectional imaging techniques, such as contrast-enhanced computer tomography (CT) and magnetic resonance imaging (MRI)
[131][132][133][370,371,372]. However, these imaging techniques are more time-consuming and less sensitive, which directs to the development of novel screening methods to detect specific biomarkers of HCC with higher sensitivity. Recently proteomics approaches have been used to detect the diagnostic and prognostic biomarkers of HCC
[134][135][136][137].[373,374,375,378]
Currently, pathological staging of tumors is considered a gold standard for CRC prognosis
[138][390]; however, it fails to predict the recurrence in patients undergoing surgical resection for colorectal cancer treatment
[138][390]. Given that some molecular mechanisms controlling colorectal carcinogenesis and its metastasis have been identified, there is a need to develop novel diagnostic and prognostic tools along with new therapies for colorectal cancer diagnosis and treatment. Proteomics-based techniques have emerged as a promising approach for the identification of prognostic and diagnostic biomarkers for CRC
[139][140][141][142][407,408,409,410].
Recently, various proteomics approaches have played an important role in the diagnosis or prognosis of leukemia. Phosphoproteomics or LC-MS/MS-based proteomics has been used for the staging of patients with AML
[143][144][145][416,417,418]. Advancements in the MS-based approaches have provided the optimized resolution for the high coverage and characterization of PTMs, and the description of the tyrosin kinome, tyrosine phophatome, and phosphotyrosine proteome is a predictive phosphorylation marker
[146][419].
Modern proteomics technologies have emerged as a new detection, management, and surveillance tool for the discovery of new biomarkers of prostate cancer. Because of the only available biomarker PSA for prostate cancer, there is an urgent need to discover new biomarkers that will lead to personalized and targeted therapies. Various proteomics approaches are being applied to investigate prostate cancer
[147][148][149][451,454,460].
Lung cancer is the most prevalent form of cancer in the world and is reported as one of the main cause of mortality. This poor survival rate is due to the delay in diagnosis resulting from the lack of early detection strategies for lung cancer
[150][464]. So, there is a necessity for identifying biomarkers for prognosis and early diagnosis of this disease. Research from recent decades has shown that proteomics studies can identify biomarkers for lung cancer
[151][152][471,472].
Over the past 20 years, advances in proteomics have allowed us to catalog, visualize, compare, and dissect patterns of proteoforms and epigenetic alterations in different forms of breast cancer tissues. These studies identify and provide insight into key drivers of oncogenic signaling, novel treatment strategies including response to therapies, and specific tumor characteristics
[12][153][154][155][17,489,491,493].
Similarly, proteomics approaches are being extensively applied in various cancer investigations.