Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Xian-Ming Chen and Version 2 by Catherine Yang.
IFN-γ is an important cytokine in both innate and adaptive immune responses during intracellular parasite infections. Elevated levels of IFN-γ is detected in both experimental animals and human patients following intracellular protozoal infections. An extensive number of studies support a protective role for IFN-γ against the infection of intracellular protozoan parasites, including Plasmodium, Toxoplasma, Cryptosporidium, Trypanosoma, Leishmania, while a few studies also indicated that IFN-γ contributes to the pathogenesis of parasite infection.
- protozoan parasites
- host defense
- interferon
- pathogenesis
- signaling pathway
1. IFN-γ Production in Protozoan Parasite Infection
IFN-γ production in intracellular protozoan-infected hosts is predominantly mediated by NK cells [1][2][3][4][5][24,25,26,27,28], and CD4+/CD8+ and Ag-specific T cells [6][7][8][9][10][29,30,31,32,33] in innate and adaptive immunity, respectively. Other types of immune cells have been reported to produce IFN-γ during protozoal infection (Figure 12); for example, natural killer T (NKT) cells were shown to secrete IFN-γ in the liver of P. yeolii-infected mice [11][12][34,35] and a significant proportion of γδ T cells and αβ T cells were reported to produce IFN-γ in the peripheral blood of Plasmodium-infected children [13][14][15][36,37,38]. CD11b+ CD45low microglia and CD11b+ CD45high blood-derived macrophages were identified as the major non-T, non-NK cells expressing IFN-γ in the brain of T. gondii-infected mice, whereas group 1 innate lymphoid cells (ILC1s) were identified to produce IFN-γ in the small intestine in response to the oral infection of T. gondii in addition to NK cells and T cells [16][17][18][39,40,41]. The production of IFN-γ by immune cells can be negatively regulated by anti-inflammatory Th2 cytokines such as IL-10 and IL-4 [19][20][21][42,43,44].
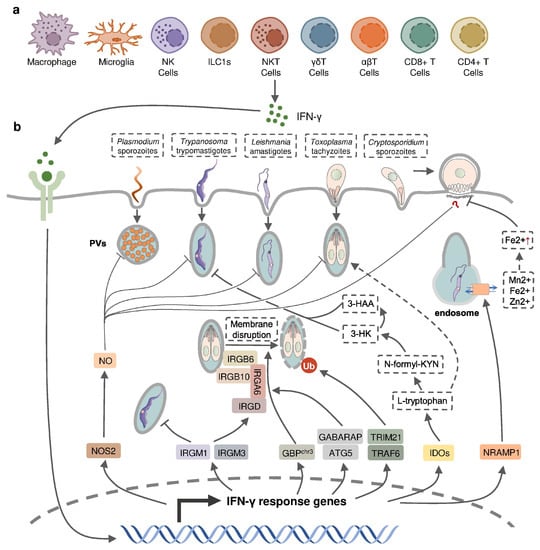
Figure 12. IFN-γ-inducible cell-autonomous defense against intracellular protozoan parasite infections. (a) IFN-γ can be produced by multiple cell types, including immune cells, during infection and (b) acts on infected host cells to eliminate intracellular parasite by nitric oxide (NO) production, the disruption of parasitophorous vacuoles (PVs) through IFN-inducible GTPase, the restriction of ion assimilation by NRAMP1, and the inhibition of nutrient acquisition by IDOs.
Table 1.
Effects of IFN-γ in host following intracellular protozoan parasite infections.
| Protective Effects | |||
| Parasite Species | Treatments and Findings | Effects | Ref. |
| P. falciparum | Recombinant IFN-γ in human hepatocyte cell culture | Hepatic schizont development ↓ | [22][45] |
| IFN-γ production in infected children | Occurrence of high-density and clinical episode of infection ↓ | [14][37] | |
| High IFN-γ level in infected patients | Occurrence of cerebral malaria ↓ | [23][46] | |
| P. berghei | Recombinant IFN-γ in rats, mice and human hepatocyte cell culture | Hepatic schizont development ↓ | [22][45] |
| Monoclonal IFN-γ neutralizing antibody in mice | Parasitemia ↑ | [24][47] | |
| Early IFN-γ production in infected mice | Occurrence of cerebral malaria ↓ | [25][48] | |
| P. vivax | Recombinant IFN-γ in chimpanzees | Parasitemia ↓ | [22][45] |
| P. Chabaudi | Recombinant IFN-γ in mice. | Parasitemia ↓ Intraerythrocytic parasites ↓ |
[22][45] |
| IFN-γ−/− mice | Parasitemia ↑ | [26][49] | |
| P. cynomolgi | Recombinant IFN-γ in rhesus monkey | Hepatic schizont development ↓ | [22][45] |
| P. yoelii | Recombinant IFN-γ in mice | Parasitemia ↓ | [27][50] |
| IFN-γR−/− mice | Infection burden ↑ | [28][51] | |
| IFN-γ−/− mice | Parasitemia ↑ | [26][49] | |
| T. b. brucei | IFN-γ−/− mice | Parasitemia ↑ Survival time ↓ |
[10][33] |
| T. b. rhodesiense | IFN-γ−/− mice | Parasitemia ↑ Survival time ↓ |
[29][52] |
| L. major | IFN-γR−/− mice | Larger and progressing lesions | [30][53] |
| L. amazonesis | IFN-γ−/− mice | Devastating lesions in late infection stages | [31][54] |
| T. gondii | IFN-γ−/− mice | Survival ↓ Infection burden ↑ |
[32][55] |
| C. parvum | Recombinant IFN-γ in intestinal enterocytes cell culture | Infection burden ↓ | [33][56] |
| IFN-γ−/− mice | Survival ↓ Occurrence of chronic infection ↑ |
[34][57] | |
| Pathogenic Effects | |||
| Parasite Species | Treatments and Findings | Effects | Ref. |
| P. berghei | Late IFN-γ production in infected mice | Occurrence of cerebral malaria ↑ | [25][48] |
| Large amount of IFN-γ produced in infected mice | Susceptibility to cerebral malaria ↑ | [35][36][58,59] | |
| IFN-γR−/− mice | No cerebral malaria development | [37][38][60,61] | |
| IFN-γ−/− mice | No cerebral malaria development | [39][40][62,63] | |
| Monoclonal IFN-γ neutralizing antibody in mice | Occurrence of cerebral malaria ↓ Survival time ↑ |
[41][64] | |
| P. yoelii, P. chabaudi, P. berghei |
Overproduction of IFN-γ | Development of Tfh and GC B cell response ↓ Humoral immunity ↓ Autoimmune anemia ↑ |
[42][43][65,66] [44][45][67,68] [46][69] |
| T. congolense | Overproducing IFN-γ in mice | Survival time ↓ | [47][70] |
| Reducing production of IFN-γ in mice | Survival time ↑ | [48][71] | |
| Monoclonal IFN-γ neutralizing antibody in mice | Susceptibility ↓ | [49][72] | |
2. IFN-γ in Host Defense against Protozoan Parasites
IFN-γ appears to be critical in controlling the infections of many intracellular parasites (Table 1). Exogenous IFN-γ was found to significantly diminish infections of Plasmodium in mice, rats, non-human primates, as well as in in vitro human hepatocytes, by inhibiting the parasite DNA replication during liver-stage development [22][45]. However, Plasmodium has also evolved a strategy to evade the host defense during liver-stage development by suppressing the expression of pro-inflammatory cytokines including IFN-γ in hepatic mononuclear cells [50][73]. Plasmodium-infected mice administrated with recombinant IFN-γ exhibited a suppressed blood-stage infection with the delayed onset of parasitemia, decreased levels of infected erythrocyte, and increased survival [27][51][52][50,74,75]. The positive effect of endogenous IFN-γ in the host defense against Plasmodium was determined from the increased susceptibility of rats treated with an anti-IFN-γ neutralizing antibody and in mice deficient in IFN-γ or the IFN-γ receptor [24][26][28][47,49,51]. Human studies also indicated a positive correlation between low IFN-γ production by live Plasmodium-stimulated peripheral blood mononuclear cells and the increased occurrence of symptomatic malaria as well as the risk of moderate-to-high-density P. falciparum reinfection [14][37]. Early IFN-γ production was shown to contribute to the protection against the development of murine cerebral malaria, the most severe neurological complication of Plasmodium infection, in P. berghei-infected mice and peripheral levels of IFN-γ were found to drop just before the onset of both human and murine cerebral malaria [23][25][46,48]. IFN-γ can be induced by malaria vaccines, as higher numbers of IFN-γ-producing T cells and increased IFN-γ level were detected in vaccine-treated subjects in several clinical trials [53][54][55][76,77,78]. Vaccination with chemically attenuated, asexual, blood-stage Plasmodium falciparum induces anti-parasitic cellular immune responses involving IFN-γ in Plasmodium-naïve volunteers [56][79]. Nevertheless, a subunit vaccine targeting Plasmodium falciparum circumsporozoite protein (CSP) activates the host immune responses dominated by parasite specific IgG antibody instead of IFN-γ [57][80]. Mice with a deficiency of IFN-γ or IFN-γ receptor have a higher susceptibility to L. major infection, accompanied by elevated Th2-type responses compared to the wild-type mice, but IFN-γ-deficient mice do not appear to succumb to L. amazonensis until 2 months post infection, suggesting that IFN-γ is induced at different stages of infection by diverse Leishmania species [30][31][58][53,54,81]. Similarly, IFN-γ or IFN-γ-receptor-deficient mice exhibited high susceptibility to infections by T. b. rhodesiense, T. b. brucei, and T. gondii [10][29][32][59][33,52,55,82]. While exogenous IFN-γ inhibits the development of C. parvum in cultured enterocytes without the need of immune effector cells, both IFN-γ-deficient and anti-IFN-γ-antibody-treated neonatal mice became more susceptible to C. parvum infection [33][34][56,57].
IFN-inducible cell-autonomous defense, including parasiticidal activity mediated by nitic oxide (NO), the disruption of parasitophorous vacuoles (PVs) related to IFN-inducible GTPase, the restriction of ion assimilation by natural resistance-associated macrophage protein 1 (NRAMP1), and the inhibition of nutrient acquisition by indoleamine 2,3-dioxygenases (IDOs), is critical for the restriction of parasite growth in infected cells and the elimination of the parasite inside the parasite-containing subcellular compartments [60][83] (Figure 12). Previous studies have underlined the role of nitric oxide synthase 2 (NOS2, iNOS)-NO in IFN-γ-mediated parasiticidal activity against intracellular protozoan parasites. IFN-γ-induced NO production showed the most evident parasiticidal activity against T. cruzi trypomastigotes and L. major amastigotes in IFN-γ-activated macrophages and P. falciparum as well as P. yoelii sporozoites in human and mouse hepatocytes, respectively [28][61][62][63][51,84,85,86]. Correspondingly, Nos2-deficient mice were highly susceptible to these pathogens [58][61][62][81,84,85]. NO production was reported to be induced by IFN-γ in hosts infected with T. gondii. Nevertheless, IFN-inducible NO might play a limited role and function at later time points in hosts infected with tachyzoites of the less virulent type II T. gondii, whereas it was essential in parasite control in virulent type I-T. gondii-strain-infected hosts [64][87]. C. parvum-infected mice had an increased level of NOS2 which was partially attributed to activated IFN-γ signaling [34][57]. A slightly longer infection period was observed in C. parvum-infected neonatal Nos2-deficient mice [65][88]. Nonetheless, several other studies with human enterocytes and mouse models indicated that the protective action of IFN-γ against C. parvum infection is independent of NO activity [33][66][67][56,89,90]. Thus, the precise mechanisms of NO-mediated antiprotozoal activity are still incompletely understood.
IFN-inducible immunity-related GTPases (IRGs) are a subfamily of IFN-γ-inducible GTPases characterized by a particular molecular weight between 21 to 47 kDa. They defend host cells against protozoa by targeting the PVs. IRGs can be divided into two groups—GKS-containing IRGs (IRGA, IRGB, IRGC or IRGD groups) and GMS-containing IRGs (IRGMs)—based on their canonical-lysine-containing (lysine, K) and non-canonical (methionine, M) G1 motifs (GxxG[K/M]S/T) within the conserved amino-terminal catalytic GTPase domain [68][91]. IRGM1, IRGM3 and IRGA6 enhance the IFN-γ-induced control of avirulent T. gondii strain in macrophages and astrocytes, which might account for the limited effect of NO in hosts infected with this parasite [69][70][71][72][73][74][92,93,94,95,96,97]. In host cells infected with avirulent T. gondii, IRGM proteins that act as guanine nucleotide dissociation inhibitors under an uninfected status are released from the endoplasmic reticulum which “turns on” the GKS-containing IRGs to target the PV [68][91]. IRGM1 also contributes to the elimination of T. cruzi infection in macrophages as IRGM1 KO macrophages displayed a defective intracellular killing of T. cruzi amastigotes [75][98]. A hierarchical model revealed the intrinsic order of IRGs when they are recruited to PV which indicated that IRGB6 and possibly IRGB10 bind to the vacuole before IRGA6, while IRGD is loaded last [76][99]. The recruitment of these molecules leads to vesiculation, membrane disruption, and sometimes necroptosis of the targeted PVs.
The other subfamily of IFN-γ-inducible GTPases, guanylate-binding proteins (GBPs), consists of a set of proteins with molecular weights between 65–73 kDa, comprising 7 and 11 members in humans and mice, respectively [77][100]. GBP genes are categorized into two clusters located on chromosome 3 and chromosome 5 in mice. Mice lacking GBPs on chromosome 3 (GBPchr3), including GBP1, GBP2, GBP3, GBP5 and GBP7, were highly susceptible to T. gondii infection due to the insufficient disruption of the PVs [78][101]. Moreover, macrophages lacking GBPchr3 showed defective loading of IRGB6 on the T. gondii PV membrane (PVM), suggesting that GBPs and IRGs coordinate with each other in PV targeting [78][101]. Compared with GBPchr3-deficient mice, mice deficient in GBP1 or GBP2 exhibited a milder decline of survival rates following T. gondii infection, indicating the complementary roles of each GBP on chromosome 3 in the host defense against parasite infection [79][80][102,103]. On the contrary, GBP1 was not recruited to T. cruzi compartments suggesting a protozoan specificity of GBP-mediated PV disruption [81][104]. The structural and biochemical cues of IFN-γ-inducible GTPase for targeting these molecules to the PV and whether membrane disruption is due to a direct effect of IRG activity, or a result of some intermediary molecules, remains unclear. Recent evidence suggests the C-terminal isoprenylation of GBP2 regulates the recruitment of GBP2 to the PVM by recognizing the ubiquitination on the PVM, which differentiates between the host membrane and the PVM [82][105]. E3 ligases such as TRAF6 and TRIM21 modulate ubiquitination of T. gondii PVM following IFN-γ treatment, whereas the dependent effect of these molecules on the IFN-γ-mediated elimination of T. gondii is controversial [83][84][85][106,107,108]. Interestingly, the distribution of GBPs in the host cytoplasm triggering the disruption of PVM is also regulated by ubiquitin-like autophagy proteins, such as autophagy-regulated gene 5 (ATG5) and GABARAP autophagy proteins, in an autophagy-independent fashion [86][87][109,110].
NRAMP1 is a highly hydrophobic integral membrane phosphoglycoprotein (~100 kD), expressed primarily in the late endosomal/early endosomal compartment of macrophages and polymorphonuclear leukocytes as a membrane transporter [88][111]. It has been shown to transport divalent ion cations, such as Mn2+, Fe2+ and Zn2+, to prevent the intracellular pathogens from these divalent metals essential for parasite survival [88][89][111,112]. NRAMP1 has been identified as an essential factor in the host defense against L. donovani, but the intrinsic mechanism remains unclear [90][113]. A previous study has revealed the correlation between cellular Fe2+ concentration and the IFN-γ-induced inhibition of C. parvum infection in intestinal enterocytes, but whether NRAMP1 is involved has not been investigated [33][56].
IDOs, IDO1 and IDO2, are both IFN-inducible, haem-containing oxidoreductases that degrade L-tryptophan to generate N-formylkynurenine (N-formyl-KYN) in the kynurenine pathway [91][114]. The removal of L-tryptophan restricted the growth of T. gondii in several IFN-γ-stimulated human cell lines including fibroblasts, lung epithelial cells, glioblastoma cells, retinal pigment epithelial cells, and macrophages [92][93][94][95][96][97][98][115,116,117,118,119,120,121]. Moreover, increased susceptibility to T. gondii occurred in mice with a double deficiency of IDO1 and IDO2 but not in IDO1-deficient mice, suggesting a significant role of both IDOs in the restriction of T. gondii infection in vivo [99][122]. IDOs may also control T. cruzi infection through the downstream kynurenine catabolites 3-hydroxykynurenine (3-HK) and 3-hydroxyanthranilic acid (3-HAA), which are likely to be harmful to T. cruzi amastigotes and trypomastigotes [100][123].
Additionally, IFN-γ could increase the expression of endothelial vascular cell adhesion molecule 1 (VCAM-1) to facilitate the recruitment of CD8+ T cells in the brain of mice chronically infected with T. gondii and enhance the cytotoxic potential of CD8+ T cells by inducing NO, which contributes to the host defense against parasites in the brain [17][101][102][40,124,125]. IFN-γ has been reported to modulate B-cell-mediated humoral immunity in Plasmodium infection via modulating the class-switching of antibody-producing B cells as IFN-γ-deficient mice produce less parasite-specific IgM, IgG3 and cytophilic IgG2a than wild-type mice [103][126].
3. IFN-γ in the Pathogenesis of Protozoan Infection
In contrast to the protective effect of IFN-γ, the response has also been reported to be involved in the pathogenesis of protozoan infection (Table 1). Although IFN-γ production as early as 24 h p.i. prevented the occurrence of experimental cerebral malaria in Plasmodium-infected mice, mice with late IFN-γ production at 3 to 4 days p.i. were found to develop severe experimental cerebral malaria [25][48]. IFN-γ mRNA accumulation was detected in mice susceptible to cerebral malaria [35][58]. The suppressed development of cerebral malaria was observed in Plasmodium-infected mice following the administration of anti-IFN-γ monoclonal antibody, treatment with IFN-γ-suppressive IL-10, inhibition of IFN-γ production, or deficiency of the IFN-γ receptor [36][37][39][41][59,60,62,64]. IFN-γ may mediate the development of experimental cerebral malaria through various mechanisms. IFN-γ, together with tumor necrosis factor (TNF) and lymphotoxin α, enhance the activation and apoptosis of the brain endothelium through the activation of endothelial cell and subsequently increased local binding of platelets [41][104][105][64,127,128]. IFN-γ is also necessary for the recruitment of CD8+ T cells in the brain by inducing the expression of canonical adhesion molecules, such as ICAM-1, CXCL9, and CXCL10. Accumulated CD8+ T cells mediate the immune responses against infected red blood cells sequestered in the brain and the lungs in susceptible mice, facilitating development of experimental CM [38][40][61,63]. The precise effect of IFN-γ in the development of cerebral malaria is still controversial. T. congolense highly susceptible BALB/c mice displayed significantly higher plasma IFN-γ levels compared to infected parasite-resistant C57BL/6 mice [49][106][107][108][72,129,130,131]. The IFN-γ-mediated accumulation and activation of erythrophagocytic myeloid cells led to acute anemia, liver injury, and a reduced survival time in T. brucei or T. congolense-infected BALB/c mice [47][48][109][70,71,132]. The overproduction of IFN-γ, induced by blocking IL-10R, shortened the survival time of both C57BL/6 and BALB/c mice following T. congolense infection [47][70]. T. congolense-susceptible BALB/c mice could switch to a relatively resistant-like phenotype by the neutralization of IFN-γ or by reducing the production of IFN-γ through the depletion of IL-12 during T. congolense infection [48][49][71,72]. Moreover, IFN-γ is also a crucial mediator in the humoral immunity that may exacerbate infection outcomes, leading to parasite-associated autoimmune disorders and chronic infection. Plasmodium DNA could induce autoreactive responses against erythrocytes by activating a population of B cells expressing CD11c and T-bet, in which IFN-γ acted as an essential factor, together with parasitic DNA to promote the expansion of autoreactive T-bet+ B cells, a major producer of autoantibodies promoting malarial anemia [46][69]. IFN-γ also contributed to the inhibition of T follicular helper cell differentiation during sever malaria infection, resulting in an impaired germ center B cell response and inefficient production of antibody-secreting plasma cells [42][43][44][45][65,66,67,68].
The precise role of IFN-γ in the host defense against protozoan parasite infection and in the pathogenesis of infection can be different during the infection of different pathogens, at different infectious stages, or in hosts with different intrinsic immune statues. Therefore, close attention to the alteration in IFN-γ levels and the IFN-γ-mediated immune response is necessary for timely adjustments of therapeutic strategies and predictions of prognosis of infection.