Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Junichi Iwata and Version 4 by Jessie Wu.
Cleft lip and palate is one of the most common congenital birth defects and has a complex etiology. Either genetic or environmental factors, or both, are involved at various degrees, and the type and severity of clefts vary. One of the longstanding questions is how environmental factors lead to craniofacial developmental anomalies.
- cleft lip
- cleft palate
- microRNA
1. microRNAs Related to Cleft Lip
As of 2022, 55 mouse genes and more than 400 human genes had been reported as related to cleft lip and palate [1][2][42,43] in the gene datasets available at CleftGeneDB (https://bioinfo.uth.edu/CleftGeneDB/index.php?csrt=15984704412663399126, accessed on 28 October 2022). Bioinformatic analysis and consequent experimental validation identified miRNA-mediated gene regulatory networks in cleft lip (Figure 1). For instance, mmu-miR-124-3p suppresses cell proliferation in cultured mouse embryonic lip mesenchymal (MELM) cells through downregulation of cleft lip-related genes Bmpr1a, Cdc42, Itf88, Pbx3, and Tgfbr1 [1][42]. In agreement with this function in MELM cells, mmu-miR-124-3p can suppress cell proliferation in other cell types, for instance, human keratinocytes (HaCaT) through FGFR2 [3][44], human non-small cell lung cancer and nasopharyngeal carcinoma cells through STAT3 [4][5][45,46], and colorectal cancer cells through PRPS1 [6][47]. Under physiological conditions in C57BL/6J mice, mmu-miR-124-3p expression in the maxillary processes (MxPs) is upregulated at embryonic day (E) E12.5 and E13.5 compared to E10.5 and E11.5 [1][42]. This suggests that miR-124-3p is expressed at very low levels during normal lip development.
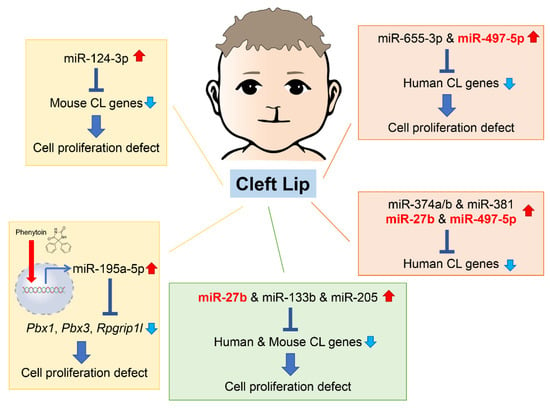
Figure 1. Summary of the microRNAs (miRNAs) and genes associated with cleft lip in humans and mice. Phenytoin is a known inducer of cleft lip in mice. It inhibits cell proliferation in cultured cells through induction of pathogenic miR-196a-5p, which suppress expression of genes related to cleft lip. CL, cleft lip.
In theour previous studies, reseaercherswe identified five miRNAs that regulate the expression of genes related to cleft lip. These miRNAs have not yet been reported or investigated in embryogenesis and craniofacial development. However, they are suggested to be associated with cancer pathogenesis and prognosis through changes in cell proliferation and differentiation. Since these miRNAs are specifically expressed under specific pathological conditions, such as cancer and cleft lip, they are considered to be pathogenic miRNAs related to cleft lip. Specifically, overexpression of hsa-miR-655-3p and hsa-miR-497-5p inhibits cell proliferation in cultured human lip mesenchymal cells through downregulation of cleft lip-related genes: BCL1, CYPLA1, DMD, FZD6, HOXB3, MID1, NTN, and SATB2 by hsa-miR-655-3p; and BAG4, CHD7, FGFR1, FOXP2, HECTD1, RUNX2, and TFAP2A by hsa-miR-497-5p [2][43]. hsa-miR-665-3p decreases cell viability by apoptosis or suppresses cell proliferation through downregulation of target genes in various cells, namely BCL2 in human lung adenocarcinoma cells [7][48], NHEG1 in human neuroblastoma [8][49], TRIM24 in human castration-resistant prostate cancer [9][50], and FZD4 in human oral squamous cell carcinoma [10][51]. In addition, hsa-miR-497-5p inhibits cell proliferation through downregulation of target genes in several human cancer cells, e.g., MAPK1 in cervical cancer cells [11][52], PDL1 or SLC7A5 in human colorectal cancer cells [12][53], and WNT3A in human nasopharyngeal carcinoma cells [13][54]. Thus, miR-124-3p, miR-655-3p, and miR-497-5p may play a key role in cell proliferation as tumor suppressors in cancers and cleft lip with/without cleft palate (CL/P) inducers in development.
Interestingly, in the miRNA, transcription factor (TF), and non-TF networks, there is a common consensus subnetwork consisting of five TF genes (GLI2, PAX3, PAX7, PAX9, and SATB2), three non-TF genes (FGFR1, RARA, and SUMO), and five miRNAs (miR-27b, miR-133b, miR-205, miR-376b, and miR-376c) in humans and mice [14][55]. In cultured human and mouse lip mesenchymal cells, miR-27b inhibits cell proliferation through gene suppression of PAX9 and RARA; miR-133b inhibits cell proliferation through gene suppression of FGFR1, PAX7, and SUMO1; and miR-205 inhibits cell proliferation through gene suppression of PAX9 and RARA [14][55]. miR-27b-3p has been reported to be a tumor suppressor, inhibiting cell proliferation and migration through target gene expression in several cancer cells: TAB3 in hepatocellular carcinoma [15][56], MLL4 in glioblastoma stem cells [16][57], TMED5 in gastric cancer cells [17][58], and CTNNB1 in ovarian endometrial cells [18][59]. Overexpression of miR-133b suppresses cell proliferation viability and migration in various cancer cells: prostatic carcinoma cells through ZNF587 [19][60] or SDCCAG3 expression [20][61], cervical cancer cells through ARFGEF1 expression [21][62], and lung adenocarcinoma through CDCA8 expression [22][63]. Interestingly, miR-133b is upregulated in the exosomes secreted from skeletal muscle cells in limb and trunk muscles during development, regulating expression of the serum response factor (SRF) and myoblast differentiation in mice [23][24][25][64,65,66]; miR-133b is also thought to contribute to lip muscle development. miR-205 suppresses cell proliferation and migration in breast cancer cells through KDM4A [26][67], glioma cells though VEGFA [27][68], and gastric cancer cells through FAM84B [28][69], and miR-205-3p is downregulated in the nucleus pulposus of the intervertebral disc, which derives from the notochord, in mouse models for intervertebral disc degeneration [29][70]. Moreover, miR-205-3p suppresses WNT/β-catenin signaling, resulting in suppression of cell proliferation and ECM synthesis [29][70].
The miRNAs described above can commonly inhibit angiogenesis through downregulation of target genes. In fact, miR-205 downregulates VEGA in gastric cancer [30][71], hepatocellular carcinoma [31][72], and the extracellular vesicles from diabetic ulcers [32][73], whereas miR133b in the exosomes secreted from bone marrow mesenchymal stem cells downregulates FBN1 [33][74] and miR-27b downregulates AMPK in brain microvascular endothelial cells [34][75], CDH5 (a.k.a. VE-cadherin) in ovarian cancer [35][76], and VEGFC in gastric cancer [36][77]. Since angiogenesis is critical for tissue growth and development, these miRNAs may play a role in various tissue processes from morphogenesis through angiogenesis.
2. microRNAs Related to Cleft Palate
An increasing number of studies show that miRNAs are involved in both normal palate and CL/P development in humans and mice (Figure 2).
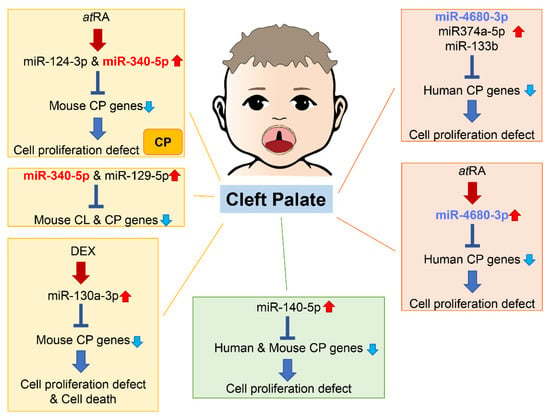
Figure 2. Summary of miRNAs and genes related to cleft palate in humans and mice. Through bioinformatic analysis for gene expression datasets and genes related to cleft palate, several miRNAs related to cleft palate are suggested to be pathogenic miRNAs. Many miRNAs among them have been validated in cultured cells and/or chemical-induced cleft palate mouse models. CL, cleft lip; CP cleft palate (including CPO and CLP).
As of 2021, 395 genes (CPO: 367 genes; anterior cleft: 16 genes; posterior/soft palate cleft: 15 genes; submucous cleft: 37 genes; and CLP: 44 genes) were reported as genes related to cleft palate in mice and 131 genes in humans [37][38][78,79]. A total of 365 mouse strains show complete cleft of the secondary palate, 44 mouse strains exhibit CLP, 14 mouse strains display anterior cleft palate, 16 mouse strains present posterior cleft palate (soft palate cleft), and 37 strains have submucous cleft palate. Overexpression of miR-374a-5p, miR-4680-3p, and miR-133b suppresses cell proliferation through the regulation of genes related to human cleft palate in cultured human palatal mesenchymal cells: ARNT, BMP2, CRISPLD1, FGFR2, JARID2, MSX1, NOG, RHPN2, RUNX2, WNT5A, and ZNF236 by miR-374a-5p; ERBB2, JADE1, MTHFD1, and WNT5A by miR-4680-3p; and FGFR1, GCH1, PAX7, SMC2 and SUMO1 by miR-133b [37][78].
Overexpression of miR-374-5p suppresses cell proliferation in several cells: in human non-small cell lung carcinoma cells by suppressing NCK1 expression [39][80], and in human neural stem cells by suppressing HES1 expression, which promotes neural stem cell differentiation [40][81]. On the other hand, miR-374-5p shows protective effects in cell viability, reducing apoptotic cell death induced by either oxygen/glucose deprivation (an infant hypoxic-ischemic encephalopathy model) in rat PC12 neuronal cells [41][82] or by LPS in human pulmonary microvascular endothelial cells [42][83]. Interestingly, maternal circulating hsa-miR-374-5p is strongly associated with the risk of small-for-gestational-age birth and preterm delivery in humans [43][44][84,85], suggesting that miR-374-5p may influence cell proliferation and survival in development.
A total of 44 CP genes are common in humans and mice. A bioinformatic analysis revealed that miR-140-5p is a potential pathogenic miRNA that specifically induces cleft palate in both humans and mice [45][86]. Overexpression of miR-140-5p suppresses genes that are crucial for palate formation (Pdgfra for the primary palate, Pax9 for the secondary palate, and Bmp2 and Fgf9 for both primary and secondary palate) in human and mouse palatal mesenchymal cells. However, the role of miR-140-5p seems to vary per cell type. Its overexpression induces adipogenic differentiation and lipogenesis through suppression of PDGFRα in pre-adipocytes [46][87] and alleviates pyroptosis by targeting Ctsb in chondrocytes treated with LPS (an osteoarthritis (OA) model) and in articular cartilage in OA mice [47][88]. On the other hand, overexpression of miR-140-5p suppresses osteogenic differentiation by targeting SATB2-mediated ERK1/2 and P38MAPK signaling pathways in human vascular smooth muscle cells [48][89]. Moreover, miR-140-5p binds to NRF2, which is a key molecule for anti-oxidative stress and cellular toxicity, enhances the NRF2/HO-1 signaling pathway, and suppresses cell proliferation, cell migration, and angiogenesis in breast cancer cells under hypoxia conditions [49][90]. In zebrafish, overexpression of miR-140 results in a cleft between lateral elements of the ethmoid plate, a structural analog of the palate in higher vertebrates, through the suppression of Pdgfra [50][91]; in mice, miR-140 null mice exhibit submucous cleft palate with hypoplastic palatal bones [51][92]. Thus, a fine-tuned, precise amount of miR-140 would be crucial for palate development. A single nucleotide polymorphism (SNP) in pre-miR-140 responsible for decreasing miR-140-5p expression is associated with an increased risk of non-syndromic CL/P (nsCL/P) in humans [52][93]. SNPs in PDGFRA are also associated with risk of developing nsCL/P, with one SNP found at the 3′-UTR near a binding site for miR-140 [53][94]. These results suggest that the miR-140–PDGFRA axis plays a crucial role in CL/P.
Mutations in TBX1 cause CL/P or CPO in humans and mice [54][55][56][95,96,97], whereas overexpression of Tbx1 suppresses Zeb2 expression in Hela cells, which induces EMT and reduces stemness [57][58][98,99], cell proliferation, and keratinocyte differentiation [59][100]. TBX1 binds to the 3′-UTR of a miR200b/200a/429 cluster (an EMT suppressor) and induces miR-200b/200a/429 expression, resulting in the suppression of Zeb2 and miR-203 in Hela and A549 cells [57][98]. miR-200b/200a/429/miR-203 negatively regulates Zeb2 expression. As expected, expression of miR-200b-5p, 429-3p, and 203-3p is significantly downregulated in palatal epithelial cells, and expression of Zeb1 and Zeb2 is upregulated in the developing palate in Tbx1 null mice. These findings suggest that the TBX1–miR-200b/200a/429 and miR-203–ZEB2 loop is important for epithelial cell differentiation, EMT, and stemness in the palatal epithelium, and their dysregulation results in CL/P. Indeed, miR-17-92 null mice (miR-17-92-/-, miR-17-9-/-;miR-106b-25+/-, and miR-17-92-/-;miR-106-/- mice) display CLP through upregulation of Tbx1, Tbx3, Fgf10, Shox2, and Osr1 expression [60][101]. On the other hand, overexpression of the miR-17-92 cluster suppresses expression of E2F1, a transcription factor, and inhibits cell proliferation through dysregulation of the cell cycle in mouse embryonic palatal mesenchymal (MEPM) cells [61][102]. Moreover, transgenic mice expressing inhibitors for miR-17-92 and miR-17-18 exhibit complete CPO through upregulation of Tgfbr1 and Tgfbr2 expression [62][103].
Human linkage analyses suggest that mutations in non-coding miRNA regions are associated with susceptibility to nsCL/P. For instance, miR-152 hypomethylation leading to overexpression is frequently detected in nsCL/P, and overexpression of miR-152 in zebrafish results in craniofacial cartilage dysmorphism [63][104]. An SNP in rs539075, located in the CDH2 intron where it is suggested to encode miRNAs, is associated with nsCL/P [64][105]. Mutations in CDH2, which plays a role in EMT, cause syndromic or non-syndromic Peters anomaly, characterized by corneal opacity, hypertelorism, and thin upper lip [65][106]. Thus, some SNPs are related to the production of miRNAs, while others are related to the binding of miRNAs. For instance, several intronic SNPs located within or near miRNA-binding sites (rs1048201/miR-496 in FGF2, rs3733336/miR-145 in FGF5, and rs546782/miR-187 in FGF9) are suggested to constitute a risk for nsCL/P [66][107]. rs12532 within the 3′-UTR of MSX1 may affect the binding to miR-3649, leading to a decrease in risk of developing nsCL/P through the regulation of MSX1 expression [67][108]. Interestingly, miR-let7-3p expression is downregulated in both the plasma from mothers carrying a nsCL/P fetus and lip tissues from nsCL/P individuals [68][109]. The inhibition of miR-let7-3p suppresses cell proliferation through HHIP upregulation and GLI2 downregulation in human oral keratinocytes. Thus, maternal miR-let-3p expression may become a potential diagnostic biomarker for nsCL/P during pregnancy. Interestingly, expression of miR-378 shows sex differences (i.e., downregulated in female nsCL/P individuals and upregulated in males) [69][110]. Increasing evidence suggests that maternal miRNA expression and SNPs in miRNA biogenesis enzymes or the 3′-UTR of CL/P-associated genes can be used for screening CL/P during pregnancy. To date, each miRNA-specific inhibitor or mimic, which can modify miRNA expression independently, is developed industrially. Several researchers have succeeded in inducing or rescuing developmental defects by administering these inhibitors/mimics to pregnant mice or zebrafishes. In the near future, these techniques can be applied to repair or reduce the severity of CL/P during pregnancy in humans.