Hepatocellular carcinoma (HCC) is one of the most prevalent cancers worldwide and the fourth leading cause of cancer-related death globally. Tumor cells recruit and remodel various types of stromal and inflammatory cells to form a tumor microenvironment (TME), which encompasses cellular and molecular entities, including cancer-associated fibroblasts (CAFs), tumor-associated macrophages (TAMs), tumor-associated neutrophils (TANs), immune cells, myeloid-derived suppressor cells (MDSCs), immune checkpoint molecules and cytokines that promote cancer cell growth, as well as their drug resistance. HCC usually arises in the context of cirrhosis, which is always associated with an enrichment of activated fibroblasts that are owed to chronic inflammation.
- cancer-associated fibroblasts
- tumor microenvironment
- tumor-associated neutrophils
- crosstalk
- hepatocellular carcinoma
1. Cellular Origin of CAFs in HCC
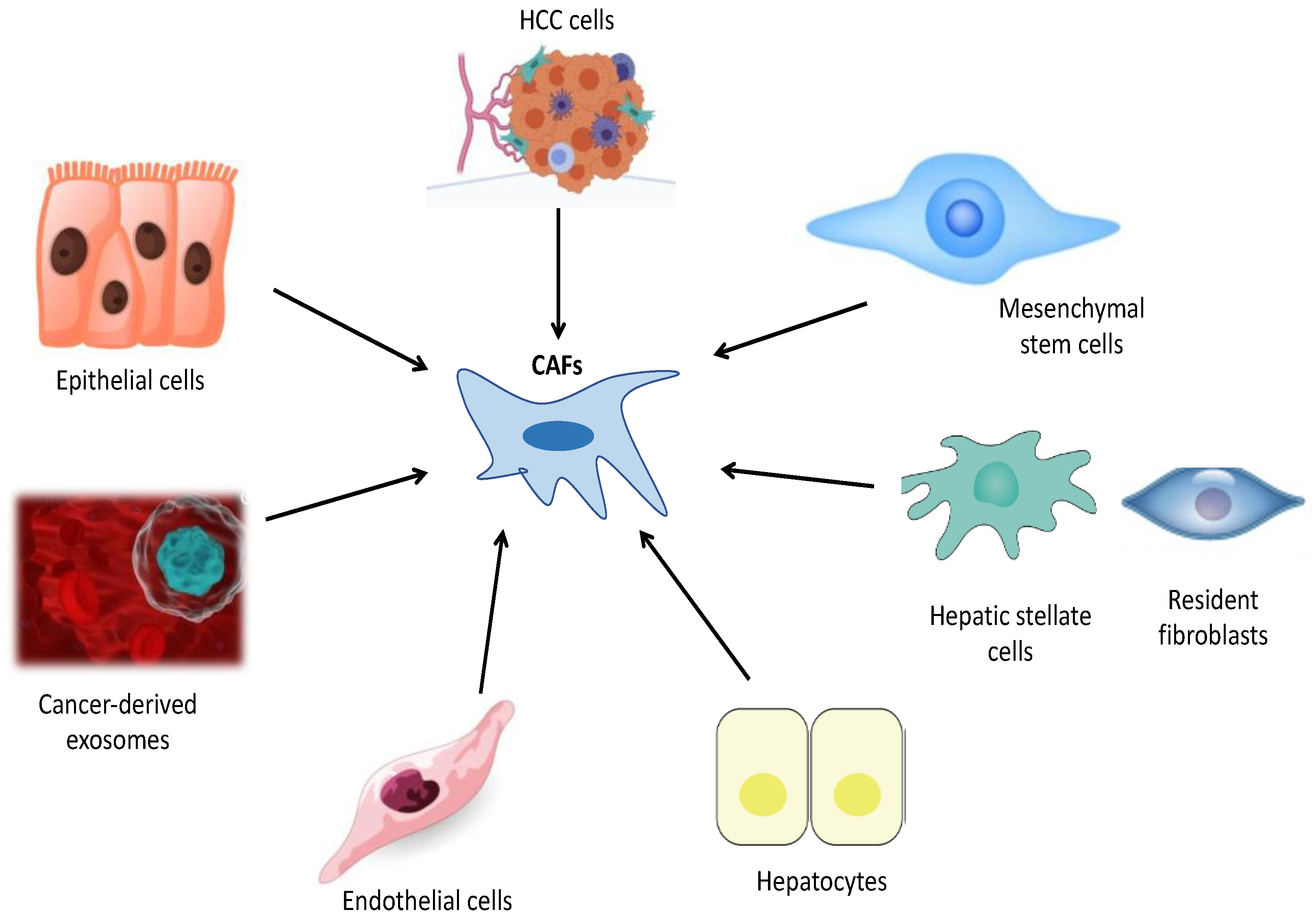
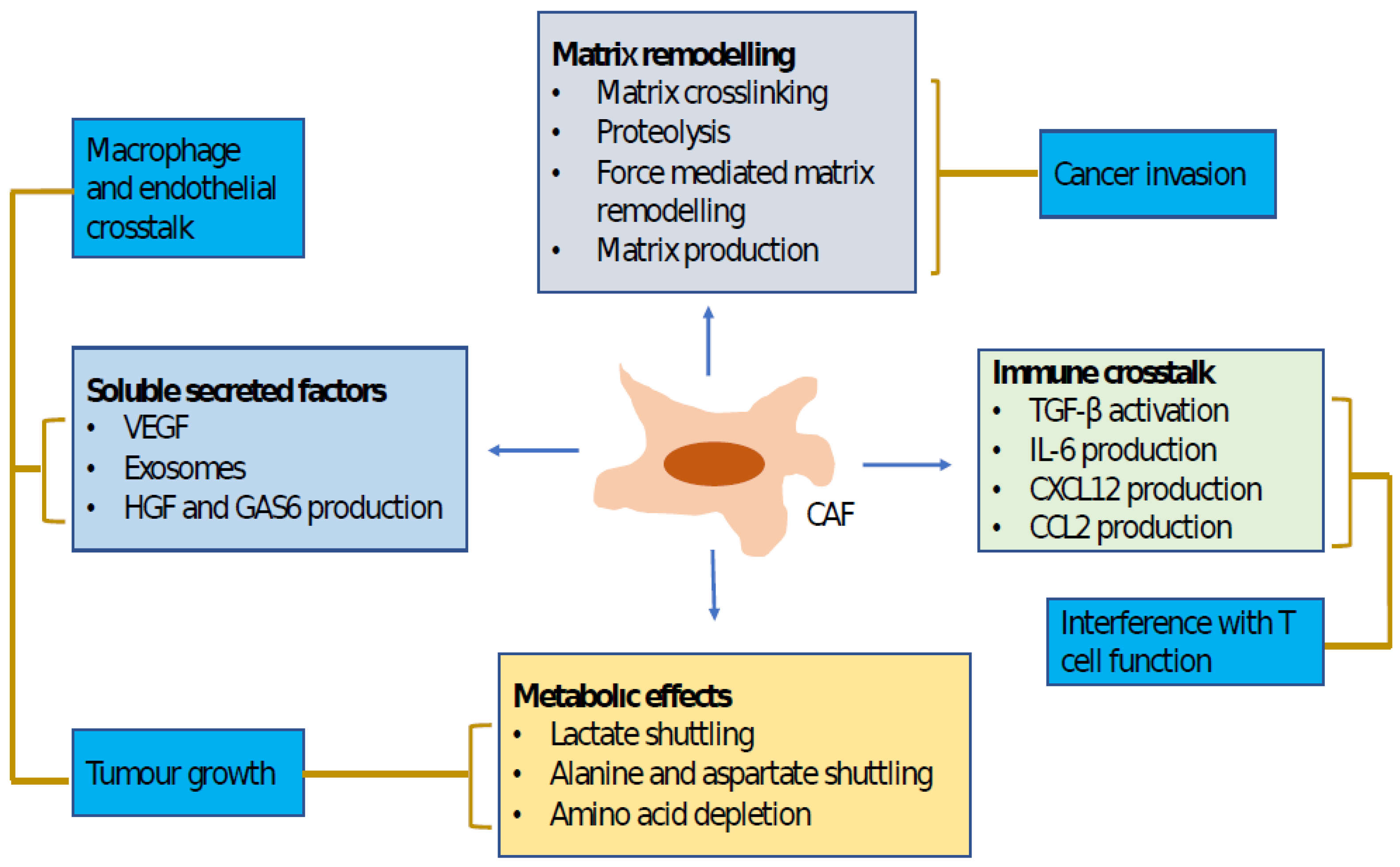
2. Impact of CAFs on HCC Progression
| Authors | Type of Trial | Signaling Pathways, Mediators and Key Findings | Reference |
|---|---|---|---|
| Mazzocca et al., 2011 | Clinical/Experimental |
|
[70][43] |
| Lau et al., 2016 | Clinical/Experimental |
|
[30][45] |
| Rhee et al., 2018 | Experimental |
|
[72][46] |
| Zhang et al., 2017 | Experimental |
|
[74][48] |
| Affo et al., 2017 | Clinical/Experimental |
|
[76][50] |
| Xu et al., 2022 | Clinical/Experimental |
|
[16][10] |
| Yang et al., 2020 | Experimental |
|
[82][56] |
| Cheng et al., 2018 | Experimental |
|
[83][57] |
| Song et al., 2021 | Clinical/Experimental |
|
[69][42] |
| Qi et al., 2022 | Experimental |
|
[63][35] |
References
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussense, L.M.; Gabrilovic, D.L.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550.
- Rix, L.L.R.; Sumi, N.J.; Hu, Q.; Deasi, B.; Bryant, A.T.; Li, X.; Welsh, E.A.; Fang, B.; Kinose, F.; Rix, U.; et al. IGF-binding proteins secreted by cancer-associated fibroblasts induce context-dependent drug sensitization of lung cancer cells. Sci. Signal 2022, 15, eabj5879.
- Biffi, G.; Tuvesan, D.A. Diversity and Biology of Cancer-Associated Fibroblasts. Physiol. Rev. 2021, 101, 147–176.
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greeten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nature 2020, 20, 174–186.
- Peng, H.; Zhu, E.; Zhang, Y. Advances of cancer-associated fibroblasts in liver cancer. Biomark. Res. 2022, 10, 59.
- Foerster, F.; Gairing, S.J.; Ilyas, S.I.; Galle, P.R. Emerging immunotherapy for HCC: A guide for hepatologists. Hepatology 2022, 75, 1604–1626.
- Peng, L.; Wang, D.; Han, Y.; Huang, T.; He, X.; Wang, J.; Ou, C. Emerging Role of Cancer-Associated Fibroblasts-Derived Exosomes in Tumorigenesis. Front. Immunol. 2022, 12, 5661.
- Zhang, J.; Gu, C.; Song, Q.; Zhu, M.; Xu, Y.; Xiao, M.; Zheng, W. Identifying cancer-associated fibroblasts as emerging targets for hepatocellular carcinoma. Cell Biosci. 2020, 10, 127.
- Mao, X.; Xu, J.; Wang, W.; Liang, C.; Hua, J.; Liu, J.; Zhang, B. Crosstalk between cancer-associated fibroblats and immune cells in the tumor microenvironment: New findings and future perspectives. Mol. Cancer 2021, 20, 131.
- Xu, H.; Zhao, J.; Li, J.; Zhu, Z.; Cui, Z.; Liu, R.; Lu, R.; Yao, Z.; Xu, Q. Cancer-associated fibroblast-derived CCL5 promotes hepatocellular carcinoma metastasis through activating HIF1α/ZEB1 axis. Nature 2022, 13, 478.
- Wang, S.-S.; Tang, X.T.; Lin, M.; Yuan, J.; Peng, Y.J.; Yin, X.; Sheng, G.G.; Ge, G.; Ren, Z.; Zhou, B.O. Perivenous Stellate Cells Are the Main Source of Myelofibroblasts Formed after Chronic liver injuries. Hepatology 2021, 74, 1578–1594.
- Bhattacharjee, S.; Hamberger, F.; Ravichandra, A.; Miller, M.; Nair, A.; Affo, S.; Filliol, A.; Chin, L.; Savage, T.M.; Yin, D.; et al. Tumor restriction by type 1 collegene opposes tumor-promoting effects of cancer-associated fibroblasts. J. Clin. Investig. 2021, 131, e146987.
- Baglieri, J.; Brenner, D.A.; Kisseleva, T. The role of Fibrosis and Liver-Associated Fibroblasts in the Pathogenesis of Hepatocellular Carcinoma. Int. J. Mol. Sci. 2019, 20, 1723.
- Ding, D.C.; Shyu, W.C.; Lin, S.Z. Mesenchymal stem cells. Cell Transpl. 2011, 20, 5–14.
- Yin, Z.; Jiang, K.; Li, R.; Dong, C.; Wang, L. Multipotent mesenchymal stromal cells play critical roles in hepatocellular carcinoma initiation, progression and therapy. Mol. Cancer 2018, 17, 178.
- Prockop, D.J. Inflammation, fibrosis, and modulation of the process by mesenchymal stem/stromal cells. Matrix Biol. 2016, 51, 7–13.
- Salah, R.A.; Nası, M.A.; El-Derby, A.M.; Elkodous, M.A.; Mohamed, R.H.; El-Ekiaby, N.; Osama, A.; Elshenawy, S.E.; Hamad, M.H.M.; Magdeldin, S.; et al. Hepatocellular carcinoma cell line-microenvironment induced cancer-associated phenotype, genotype and functionality im mesenchymal stem cells. Life Sci. 2022, 288, 120168.
- Bhattacharya, M.Z.; Kim, V.M.; Guo, H.; Talbot, L.J.; Kuo, P.J. Osteopontin promotes CCL5-mesenchymal stromal cell-mediated breast cancer metastasis. Carcinogenesis 2011, 32, 477–487.
- Yeon, J.H.; Jeong, H.E.; Seo, H.; Cho, H.; Kim, K.; Na, D.; Chung, S.; Park, J.; Choi, N.; Kang, J.Y. Cancer-derived exosomes trigger endothelial to mesenchymal transition followed by induction of cancer-associated fibroblasts. Acta Biomater. 2018, 76, 146–153.
- Greening, D.W.; Gopal, S.K.; Mathias, R.A.; Liu, L.; Sheng, J.; Zu, H.-J.; Simpson, R.J. Emerging roles of exosomes during epithelial-mesenchymal transition and cancer progression. Semin. Cell. Dev. Biol. 2015, 40, 6071.
- Zeisberg, M.; Yang, C.; Martino, M.; Duncan, M.; Rieder, F.; Tanjore, H.; Kalluri, R. Fibroblasts Derive from Hepatocytes in Liver Fibrosis via Epithelial to Mesenchymal Transition. J. Biol. Chem. 2007, 282, 23337–23347.
- Taura, K.; Miura, K.; Osterreicher, C.H.; Kodama, Y.; Penz-Osterreicher, M.; Brenned, D.A. Hepatocytes do not undergo epithelial-mesenchymal transition in liver fibrosis in mice. Hepatology 2010, 51, 1027–1036.
- Zou, B.; Liu, X.; Zhang, B.; Gong, Y.; Cai, C.; Li, P.; Chen, J.; Xing, S.; Chen, J.; Peng, S.; et al. The expression of FAP in hepatocellular carcinoma cells is induced by hypoxia and correlates with poor clinical outcomes. J. Cancer 2018, 9, 3278–3286.
- Wu, F.; Yang, J.; Liu, J.; Wang, Y.; Mu, J.; Zeng, Q.; Deng, S.; Zhou, H. Signaling pathways in cancer-associated fibroblasts and targeted therapy for cancer. Signal Transduct. Target. Ther. 2021, 6, 218.
- Cathy, J.M.; Gamarouidi, F.S.; Lio, Z.; McManus, B.M. Wnt3a induces myofibroblasts differentiation by upregulating TGF-β signaling through SMAD2 in a β-catenin-dependent manner. PLoS ONE 2011, 6, 19809.
- Jakobson, T.; Treuter, E.; Gustafsson, J.A.; Steffensen, K.R. Liver X receptor biology and pharmacology: New pathways, challenges and opportunities. Trend. Pharmacol. Sci. 2012, 33, 394–404.
- Calkin, A.C.; Tontonoz, P. Transcriptional integration of metabolism by the nuclear sterol-activated receptors LXR and FXR. Nat. Rev. Mol. Cell. Biol. 2012, 13, 213–224.
- Moren, A.; Bellomo, C.; Tsubakihara, Y.; Kardasis, D.; Mikulits, W.; Heldin, C.-H.; Moustakas, A. LXRα limits TGFβ-dependent hepatocellular carcinoma associated fibroblast differentiation. Oncogenesis 2019, 36, 36.
- Wang, H.; Feng, L.; Lu, M.; Zhang, B.; Xu, Y.; Zeng, Q.; Xi, J.; Zhou, J.; Ying, X.; Zhang, J.; et al. Integrative single-cell transcriptome analysis reveals a subpopulation of fibroblasts associated with favorable prognosis of liver cancer patients. Transl. Oncol. 2021, 14, 100981.
- Bhowmick, E.G.; Neilson, E.G.; Moses, H.L. Stromal fibroblasts in cancer initiation and progression. Nature 2004, 432, 332–337.
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598.
- Hu, J.; Li, J.; Fu, M.; Zhao, X.; Wang, W. The JAK/STAT signaling pathway: From bench to clinic. Signal Transduct. Target. Ther. 2021, 6, 402.
- Liu, J.; Chen, S.; Wang, W.; Ning, B.F.; Chen, F.; Shen, W.; Xie, W.-F.; Zhang, X. Cancer-associated fibroblasts promote hepatocellular metastasis through chemokine-activated hedgehog and TGFβ pathways. Cancer Let. 2016, 379, 49–59.
- Yu, L.; Shen, N.; Shi, Y.; Shi, X.; Fu, X.; Li, S.; Zhu, B.; Yu, W.; Zhang, Y. Characterization of cancer-related fibroblasts (CAFs) in hepatocellular carcinoma and construction of CAF-based risk signature based on single-cell RNA-seq and bulk RNA-seq data. Front. Immunol. 2022, 13, 1009789.
- Qi, Y.; Wang, H.; Zhang, Q.; Liu, Z.; Wang, T.; Wu, Z.; Wu, W. CAF-Released Exosomal miR-20-a-5p Facilities HCC Progression via the LIMA-1Mediated β-Catenin Pathway. Cells 2022, 11, 3857.
- Chung, J.Y.-F.; Chan, M.K.-K.; Li, J.S.-F.; Chan, A.S.-W.; Tang, P.C.-T.; To, K.-F.; Lan, H.-Y.; Tang, P.M.-K. TGF-β Signaling: From Tissue Fibrosis to Tumor microenvironment. Int. J. Mol. Sci. 2021, 22, 7575.
- Jangwirth, U.; van Wevarjik, A.; Evans, R.J.; Jenkins, L.; Vicente, D.; Alexander, J.; Gao, Q. Impairment of a distinct cancer-associated fibroblast population limits tumor growth and metastasis. Nat. Commun. 2021, 12, 3536.
- Ohlund, D.; Elyada, E.; Tuveson, D. Fibroblast heterogeneity in the cancer wound. J. Exp. Med. 2014, 211, 1503–1523.
- Ohlund, D.; Handly-Santana, G.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvisa, M.; Corbo, V.; Oni, T.A.; Hearn, S.A.; Lee, E.J. distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596.
- Lambrechts, D.; Wauters, F.; Boeckx, B.; Albar, S.; Nittner, D.; Burton, O.; Bassez, A.; Decaluwe, H.; Pircher, A.; van den Eynde, K.; et al. Phenotype molding of stromal cells in the lung tumor microenvironment. Nat. Med. 2018, 24, 1277–1289.
- Chiavarina, B.; Ronca, R.; Otaka, Y.; Sutton, R.B.; Rezzola, S.; Yokobori, T.; Chiodelli, P.; Souche, R.; Pourquier, D.; Yokobori, T.; et al. Fibroblast-derived prolargin is a tumor suppressor in hepatocellular carcinoma. Oncogene 2022, 41, 1410–1420.
- Song, M.; He, J.; Pan, Q.-Z.; Yang, J.; Zhao, J.; Zhang, Y.-J.; Huang, Y.; Tang, Y.; Wang, Q.; He, J.; et al. Cancer-Associated Fibroblasts-Mediated Cellular Crosstalk Supports Hepatocellular Carcinoma Progression. Hepatology 2021, 73, 1717–1735.
- Mazzocca, A.; Dituri, F.; Lupo, L.; Quaranta, M.; Antonaci, S.; Giannelli, G. Tumor-secreted Lysophostatidic Acid Accelerates Hepatocellular Carcinoma Progression by Promoting Differentiation of Peritumoral Fibroblasts in Myofibrolbasts. Hepatology 2011, 54, 920–930.
- Lei, M.M.L.; Lee, T.K.W. Cancer-Associated Fibroblasts: Orchestrating the Crosstalk Between Liver Cancer Cells and Neutrophils Through the Cardiotrophin-Like Cytokine Factor1-Mediated Chemokine (C-X-C motif) Ligand 6/TGF-β Axis. Hepatology 2021, 5, 1631–1633.
- Lau, E.Y.T.; Lo, J.; Cheng, B.Y.L.; Ma, M.K.F.; Lee, J.M.F.; Ng, J.K.Y.; Chai, S.; Lin, C.H.; Tsang, S.Y.; Ma, S.; et al. Cancer-Associated Fibroblasts Regulate Tumor-Initiating Cell Plasticity in Hepatocellular Carcinoma through c-Met/FRA1/HEY1 Signaling. Cell Rep. 2016, 15, 1175–1189.
- Rhee, H.; Kim, H.-Y.; Choi, J.-H.; Woo, H.G.; Yoo, J.E.; Nahm, J.H.; Choi, J.-S.; Park, Y.N. Keratin 19 Expression in Hepatocellular Carcinoma Is Regulated by Fibroblast-Derived HGF via a MET-ERK1/2-AP1 and SP1 Axis. Cancer Res. 2018, 78, 1619–1631.
- Yin, Z.; Dong, C.; Jiang, K.; Xu, Z.; Li, R.; Guo, K.; Shao, S.; Wang, L. Heterogeneity of cancer-associated fibroblasts and roles in the progression, prognosis, and therapy of hepatocellular carcinoma. J. Hematol. Oncol. 2019, 12, 101–122.
- Zhang, Z.; Li, X.; Sun, W.; Yue, S.; Yang, J.; Li, J.; Ma, B.; Wang, J.; Yang, X.; Pu, M.; et al. Loss of exosomal miR-320a from cancer-associated fibroblasts contributes to HCC proliferation and metastasis. Cancer Lett. 2017, 397, 33–42.
- Gascard, P.; Tisty, T.D. Carcinoma-associated fibroblasts: Orchestrating the composition of malignancy. Genes Dev. 2016, 30, 1002–1019.
- Affo, S.; Yu, L.X.; Schwabe, R.F. The Role of Cancer-Associated Fibroblasts and Fibrosisin liver Cancer. Annu. Rev. Pathol. 2017, 12, 153–186.
- Boissonnas, A.; Combadiere, C. Modulating the tumor-associated macropahge landscape. Nat. Cancer Immunol. 2022, 23, 472–482.
- Lin, Z.Y.; Chuang, H.Y.; Chuang, W.L. Cancer-associated fibroblasts up-regulate CCL2, CCL26, IL6 and LOXL2 genes related promotion of cancer progression in hepatocellular carcinoma cells. Biomed. Pharmacother. 2012, 66, 525–529.
- Kamoub, A.E.; Dash, A.B.; Vo, A.P.; Sulluvian, A.; Brooks, M.W.; Bell, G.W. Mesenchymal stem cells within tumor stroma promote breast cancer metastasis. Nature 2007, 449, 557–563.
- Liu, T.; Han, C.; Wang, S.; Fang, P.; Ma, Z.; Xu, L.; Yin, R. Cancer-associated fibroblasts: An emerging target of anti-cancer immunotherapy. J. Hemotol. Oncol. 2019, 12, 86.
- Chen, S.; Morine, Y.; Tokuda, K.; Yamada, S.; Saito, Y.; Nishi, M.; Ikemoto, T.; Shimada, M. Cancer-associated fibroblast-induced M2-polarized macrophages promote hepatocellular carcinoma progression via the plasminogen activator inhibitör-1 pathway. Int. J. Oncol. 2021, 59, 1–12.
- Yang, F.; Wei, Y.; Han, D.; Li, Y.; Shi, S.; Jiao, D.; Wu, J.; Zhang, Q.; Shi, C.; Song, W.; et al. Interaction with CD68 and regulation of GAS6 Expression by Endosialin in Fibroblasts Drives Recruitment and Polarization of Macrophages in Hepatocellular Carcinoma. Cancer Res. 2020, 80, 3892–3905.
- Cheng, Y.; Li, H.; Deng, Y.; Tai, Y.; Zeng, K.; Zhang, Y.; Liu, W.; Zhang, Q.; Yang, Y. Cancer-associated fibroblasts induce PDL1+ neutrophils through the IL-6/STAT3 pathway that foster immune suppression in hepatocellular carcinoma. Cell Death Dis. 2018, 9, 422–434.
- Ringelhan, M.; Pfister, D.; O’Connor, T.; Pikarsky, E.; Heikenwalder, M. The immunology of hepatocellular carcinoma. Nat. Immunol. 2018, 19, 222–232.
- Rizvi, S.; Wang, J.; El- Khoueiry, A.B. Liver Cancer Immunity. Hepatology 2021, 73, 86–103.
- Fu, Y.; Liu, S.; Zeng, S.; Shen, H. From bench to bed: The tumor immune microenvironment and current immunotherapeutic strategies fro hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2019, 38, 396.
- Sangro, B.; Sarobe, P.; Hervas-Stubbs, S.; Melero, I. Advances in immunotherapy for hepatocellular carcinoma. Nat. Rev. Gastronterol. Hepatol. 2021, 18, 525–543.
- Miranda, A.; Hamilton, P.T.; Zhang, A.W.; Pattnaik, S.; Becht, E.; Mezheyeuski, A.; Bruun, J.; Micke, P.; de Reynies, A.; Nelson, B.H. Cancer stemness, intratumoral heterogeneity, and immune response across cancers. Proc. Natl. Acad. Sci. USA 2019, 116, 9020–9030.
- Arvanitakis, K.; Mitroulis, I.; Germanidis, G. Tumor-associated neutrophils in hepatocellular carcinoma pathogenesis, prognosis, and therapy. Cancers 2021, 13, 2899.
- He, G.; Zhang, H.; Zhou, J.; Wang, B.; Chen, Y.; Kong, Y.; Xie, X.; Wang, X.; Fei, R.; Wei, L.; et al. Peritumoral neutrophils negatively regulate adaptive immunity via the PD-L1/PD-1 signaling pathway in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2015, 34, 141–152.
- Jakobetz, M.A.; Chan, D.S.; Neese, A.; Bapiro, T.E.; Cook, N.; Freese, K.K.; Feig, C.; Nakagawa, T.; Caldwell, M.E.; Zecchini, H.I.; et al. Hyaluronan impairs vascular function and drug delivery in a Mouse model of pancreatic cancer. Gut 2013, 62, 112–120.