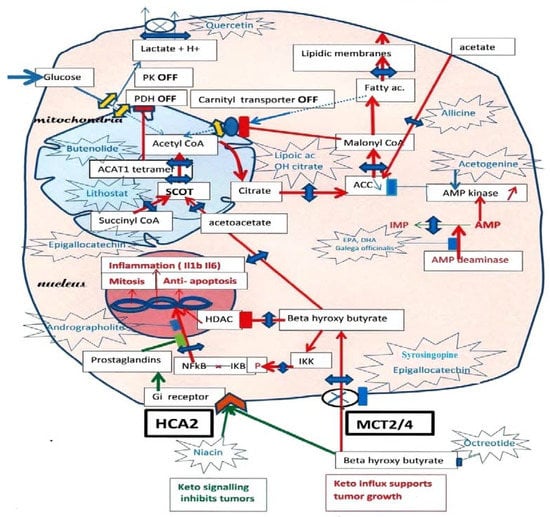
In tumor cells, ketolysis “via” succinyl-CoA: 3-oxoacid-CoAtransferase (SCOT) and acetyl-CoA acetyltransferase 1 (ACAT1) is a major source of mitochondrial acetyl-CoA. Active ACAT1 tetramers stabilize by tyrosine phosphorylation, which facilitates the SCOT reaction and ketolysis. Tyrosine phosphorylation of pyruvate kinase PK M2 has the opposite effect, stabilizing inactive dimers, while pyruvate dehydrogenase (PDH), which is already inhibited by phosphorylation, is acetylated by ACAT1 and is doubly locked. This closes the glycolytic supply of acetyl-CoA. In addition, since tumor cells must synthesize fatty acids to create new membranes, they automatically turn off the degradation of fatty acids into acetyl-CoA (“via” the malonyl-CoA brake for the fatty acid carnityl transporter). Thus, inhibiting SCOT the specific ketolytic enzyme and ACAT1 should hold back tumor progression. However, tumor cells are still able to take up external acetate and convert it into acetyl-CoA in their cytosol “via” an acetyl-CoA synthetase, which feeds the lipogenic pathway; additionally, inhibiting this enzyme would make it difficult for tumor cells to form new lipid membrane and survive.
1. A Possible Reset of the Metabolic Rewiring Mechanism in Cancer
Tumor cells avidly consume glucose, but two bottlenecks at
pyruvate kinase M2 (PK M2
) and pyruvate dehydrogenase (PDH) and PDH cut the entry in oxidative metabolism. This is similar to the process that occurs if a phosphorylation inhibiting these enzymes does not reverse while switching from neoglucogenesis to glycolysis. If one triggers neoglucogenesis through a period of fasting, in which these phosphorylations normally occur, one may try to reset the system at the interruption of the fasting period in the hope that the phosphatases will correct the anomaly, which maintains them in the OFF configuration. This switching mechanism, involving the role of calcium, a PDE and a phosphatase such as calcineurin, was discussed earlier in the paper. During the fasting period, one should accept complements that preserve calcium stores and the enzymes capable of resetting the system. On the other hand, fasting forms ketone bodies that feed the tumor;
researcherswe have indicated that these fasting ketones are not as bad as are those from high fat ketogenic diets. The incorporation of complements such as green tea and epigallocatechin
[1][2][33,34] during the fasting period would inhibit MCT2-4 transporters and the ketone influx while favoring the ketone signaling action transmitted by HCA2 receptors, which is helpful. Moreover, DHA and EPA would limit the ketone contribution to the lipogenic pathway. At the end of a long fasting period, one observes some favorable cases; they might result from a successful reset, removing the abnormal phosphorylation blocking the enzymes PK, M2 and PDH.
2. Cutting the Ketone Influx
In
Figure 1,
reswe
archers recall the inhibition of both the glycolytic and fatty acid sources of mitochondrial acetyl-CoA indicated by PK and PDH interruptions on the glycolytic entry, while a single interruption by malonyl-CoA cuts the fatty acid source of acetyl-CoA, since the tumor must synthesize its fatty acids to create new membranes. Hence, the ketolysis of BHB becomes a major acetyl-CoA supply, promoting, for example, the growth of breast tumors. Adipocytes form BHB, which enters tumor cells through MCT 2 transporters
[3][4][35,36].
Figure 1. Ketolytic dependent tumor cells. Inhibitions by phosphorylation, of Pyruvate kinase PK M2, and Pyruvate dehydrogenase PDH, cut the glycolytic supply of acetyl-CoA to mitochondria, lactic acid release occurs through monocarboxylic acid transporter (MCT) monocarboxylate transporters. Moreover, an acetylation of PDH by ACAT1 thiolase further increases the inhibition of PDH. Since tumors must synthesize fatty acid to create new membranes, this automatically turns off the fatty acid mitochondrial carnityl transporter and the fatty acid supply of acetyl-CoA. The carnityl transporter is inhibited by malonyl-CoA, the product of Acetyl CoA carboxylase (ACC), starting the lipogenic pathway in the cytosol. Hence, ketone bodies and beta-hydroxybutyrate, (BHB) become an essential source of acetyl-CoA in tumors, via SCOT the specific ketolytic enzyme and ACAT1. Then, citrate exits the mitochondria and feeds lipogenesis via ATP citrate lyase (ACL) and acetyl-CoA carboxylase (ACC). By blocking the MCT transporters with indicated compounds, one would affect the lactate release, the influx of beta-hydroxybutyrate (BHB) and the supply of acetoacetate to SCOT. ResearchersWe indicate possible inhibitors for SCOT: Lithostat or other Siderophores, Ferrichrome, Phycocyanin, Febrifugine or even Melatonin. As for ACT1, Butenolides, vitamin C and others are indicated; this should starve the tumor. ResearchersWe suggest inhibiting the citrate condensation and supply to the lipogenic pathway using lipoic acid and hydroxycitrate, as indicated. Moreover, Allicine would cut the direct conversion of acetate into acetyl-CoA in the cytosol. An essential key is that AMP kinase will inhibit ACC, which merits stimulation by acetogenin, and to inhibit AMP deaminase with Docosahexaenoic acid (DHA) or Galega officinalis, or metformin. By inhibiting the entry of BHB, one also stops the stimulation of NFkB-mediated transcription, a hallmark of cancer, and cancels the inhibition of histone deacetylase (HDAC) caused by BHB. Upstream of ketolysis, one may use octreotide (a somatostatin analog) to decrease the ketogenic supply. There is, however, a paradox, since the signaling action of extracellular BHB on HCA2, a Gi coupled receptor, is to preserve (it inhibits NFkB transcription). A prostaglandin messenger is involved; it can be strengthened with Andrographolite. Niacin is another agonist for HCA2, which may help when using octreotide for decreasing ketogenesis. Green arrows: HCA2 signaling. Red arrows: BHB influx through MCT and effects.
One should also control the BHB influx. Indeed, once in the cell, BHB has multiple effects; first, it feeds the SCOT pathway; second, it goes to the nucleus, where it inhibits HDAC, favoring the acetylation of histones, which induces the expression of fetal genes. A third effect of intracellular BHB is the activation of NFkB-mediated transcription of inflammatory cytokines Il1B. Indeed, intracellular BHB induces phosphorylation of IKB, liberating the NFkB transcription factor, which moves to the nucleus. Thus, if one inhibits the influx of BHB with compounds such as Syringopine or Syrosingopine from Rauwolfia
[4][36] or others such as Quercetin or Epigallocatechin
[1][2][33,34] tumor cells will starve, since ketolysis should decline, particularly if associated with SCOT and ACAT1 inhibitors. In parallel, the decrease in intracellular BHB stops the inhibition over HDAC, which will deacetylate histones, silencing fetal genes, and elicit a transition to adult genes. Hydroxamic acid HDAC derivatives, which display anticancer effects, are probable SCOT inhibitors, and may be able to starve the tumor. Finally, the decrease in intracellular BHB stops the stimulation of inflammatory cytokines transcription mediated by NFkB, since NFkB remains bound to IkB in the cytosol.
In summation, the MCT transporter of BHB driven by SCOT-ACAT1 attracts this essential nutrient for tumor cells. In parallel, this elicits NFkB transcription and inflammation, whereas HDAC inhibition by BHB elicits histone acetylation and the expression of fetal genes. By inhibiting the MCT transporter of BHB and the SCOT-ACAT1 pathway, one starves the tumor and blocks NFkB transcription; histones are deacetylated by HDAC, favoring a shift to adult genes. Moreover, extracellular BHB activates HCA2 receptor signaling as other agonists of these receptors, such as niacin. These receptors inhibit inflammation by keeping NFkB bound to IkB in the cytosol, further blocking inflammation and inflammatory macrophages; these effects would be mediated by prostaglandins. Niacin or other agonists of this receptor should decrease inflammation and NFkB-mediated transcription, as adrographolide (Andrographis Paniculata)
[5][37].
In conclusion, the ketone body is favorable if it stays outside the cell, acting as a signaling molecule rather than as a nutrient for the tumor. This summarizes the keto paradox and explains some opposing observations on the effects of BHB on tumor cells.
3. Cutting the Succinyl-CoA: 3-Oxoacid-CoAtransferase (SCOT)- Acetyl-CoA Acetyltransferase OT-ACAT1 Ketolytic Steps
Our strategy is to block the ketolytic pathway, particularly the specific step driven by SCOT and the supply of acetyl-CoA to lipogenesis, as
reswe
archers recently attempted on an animal cancer model
[6][38]. Moreover, since active ACAT1 tetramers collaborate with SCOT to pull in the ketolytic nutrient,
rwe
searchers suggest using inhibitors of both enzymes.
A typical inhibitor for SCOT is acetohydroxamic acid, which was introduced by Pickart and Jencks
[7][39]. There are many hydroxamic acid derivatives in the Siderophores iron chelator family molecules, and it would be interesting to test them on SCOT activity. An association with ACAT1 inhibitors, Butenolides, including vitamin C, Arecoline and trimetazidine (Vastarel) discussed above would certainly be interesting to test.
4. Possible Compounds for Inhibiting Succinyl-CoA: 3-Oxoacid-CoAtransferaseOT
Lithostat acetohydroxamic acid is a typical SCOT inhibitor used to treat bladder stones. Another inhibitor, Pimozide
[8][9][40,41], used to treat mental diseases, reduces cancer incidence. Several interesting compounds are highlighted in the work of Lissanti’s group, who describe potential SCOT inhibitors, the Mitoketoscine
[10][42], through their structural and binding properties. Selecting the best and least toxic derivative for animal cancer models requires collaboration with pharmaceutical groups.
We can also explore the literature for hydroxamic acid derivatives given for other pathologies; this is the case for Vorinostat SAHA, or Quisinostat and Trichostatin, which are HDAC inhibitors
[11][43] that display anticancer properties; it would be interesting to see if they inhibit SCOT as Lithostat. Another way to find potential SCOT inhibitors comes from bacteriology. In order to survive in a hostile environment such as in stomach acid or in the bladder, the bacteria Helicobacter or Proteus use their urease to make ammonia and neutralize the acidity. Drugs such as acetohydoxamic acid inhibit the urease by binding the metal (Ni) in the active center, blocking the alkalinization of urine and stone formation. Another source of compounds used to find SCOT inhibitors is the pharmacology of Metalloproteinase inhibitors; again, the hydroxamate derivatives that block the Zn-metalloproteinases deserve to be studied in relation to SCOT inhibition
[12][44]. The iron-binding siderophores of bacteria, algae and other organisms are likely the best to explore. In our aerobic environment, iron bioavailability is low due to its insolubility. Iron is necessary for the survival of these organisms; thus, they synthesize iron chelators to capture ferric iron. One category of these siderophores is hydroxamate derivatives; they are potential SCOT inhibitors and have clear anticancer properties
[13][45].
ResWe
archers recently compared acetohydroxamic Lithostat and Lithothamnion from red algae Lithothamnion calcareum
[14][46] on the Lewis cancer model and confirmed observations on their anticancer properties
[6][38]. An interesting siderophore is Ferrichrome from Lactobacillus casei, which inhibits colon cancer
[15][47]. The iron chelators desferroxamine (desferal) display antineuroblastoma effects
[16][48], and as such they would be interesting for future leukemia treatments. Soluble seawater marine siderophores (Ferrioxamines) phytoplankton siderophore from Rhodomonas Ovalis or siderophore from the marine bacterium Alteromonas luteoviolacea (alterobactine)
[17][49] would be interesting to test using the SCOT assay developed by Williamson
[18][19][50,51]. The problem is finding the best SCOT inhibitor that would reach the enzyme without being toxic. Presumably, a hydroxamate derivative originating from the biology of siderophores or a compound related to iron metabolism would do the job.
ResearchWe
rs know that the normal liver never expresses the specific ketolytic enzyme SCOT; only ketogenesis takes place in liver. Is this related to the recycling of the heme, iron recovery and biliverdin, bilirubin metabolism in the liver?
During bacteria or parasite infections, the microorganisms try to rob our iron with their siderophores, and proteins of our immune system with better affinity for iron compete with bacterial siderophores.
A colleague pointed out that melatonin has reactive groups similar to acetohyroxamic acid.
RWe
searchers also notice that febrifugine from Dichoa febrifuga used in Chinese medicine and its derivative Halofuginone
[20][52] have similar reactive radicals; they both have anticancer effects, affinity for iron and might inhibit SCOT.
5. Compounds Affecting Acetyl-CoA Acetyltransferase AT1
ACAT1 activity in association with SCOT secures the vital ketolytic supply to tumor cells by pulling in the influx of BHB. The antifouling Butenolide, or Vitamin C, or Karrikins and strigol from smoke, would inhibit ACAT1. Arecoline from Areca catechu is an ACT1 inhibitor, but it might have oral carcinogenic effects. Other ACAT1 inhibitors (Vastarel) could be tested in this context in association with SCOT inhibitors. The acetogenin from anones are particularly interesting, since they have a furan ring. As butenolide, they might inhibit ACAT1 and inhibit AMP deaminase, which stimulates AMP kinase that cuts the lipogenic supply. Fungal siderophores from Phellinus linteus or Phellinus baummii
[21][22][53,54] are identified, and might influence SCOTand ACAT1, inoscavin has a furan ring, which likely affects ACAT1.
6. Compounds Acting down Stream of Succinyl-CoA: 3-Oxoacid-CoAtransferase (SCOT)- Acetyl-CoA Acetyltransferase OT-ACAT1
In earlier works
rwe
searchers showed that Lipoic acid and Hydroxycitrate from Garcinia were able to limit the citrate efflux from mitochondria and inhibit ATP citrate lyase (ACL) in the cytosol, which cuts the lipogenic supply. These effects are strengthened if one uses allicine or orotic acid to block the direct incorporation of external acetate via acetyl-CoA synthetase. Moreover, one can impair the synthesis of lipid membranes with DHA through the inhibition of AMP deaminase, which leaves more AMP to stimulate AMP kinase. This then inhibits ACC, at the start of the lipogenic pathway. One can reinforce these effects by decreasing the synthesis of cholesterides, using statins that are difficult to handle, or Bergamotin
[23][55]. An interesting report
[24][56] shows that tumors can use the glutamine entry in the citric acid cycle to form citrate and feed the lipid synthesis pathway. The role of isocitrate dehydrogenase (IDH2) is essential in this process; IDH2 mediates a reductive carboxylation to facilitate the utilization of glutamine through alpha ketoglutarate to citrate reversible steps of the Krebs cycle and feed the citrate supply to lipid synthesis.
TIn this report, the long-term induction of the oncogene K-ras upregulates IDH2; this facilitates glutamine utilization for lipid synthesis during malignant transformation.
There is certainly a great variability among tumors and their metabolic adaptations. The presentation given in this resviearchw did not consider the different tumor situations or the magnitude of the glycolytic bottlenecks, or the degree of the ketone dependency.