Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Catherine Yang and Version 1 by Patrice Marques.
Non-alcoholic fatty liver disease (NAFLD) is currently the most prevalent chronic liver disease in Western countries, affecting approximately 25% of the adult population. This condition encompasses a spectrum of liver diseases characterized by abnormal accumulation of fat in liver tissue (non-alcoholic fatty liver, NAFL) that can progress to non-alcoholic steatohepatitis (NASH), characterized by the presence of liver inflammation and damage. Lymphocytes are certainly the most relevant leukocytes of the adaptive immune system.
- NAFLD
- steatosis
- inflammation
- leukocytes
1. Neutrophils
Neutrophils are the most abundant leukocytes in human blood and the first line of immune defense against infection or injury, contributing to the acute inflammatory response. This leukocyte subset is continuously released from the bone marrow, and their release into the peripheral blood is tightly regulated by different molecules, including granulocyte colony-stimulating factor (G-CSF), and ligands of CXC chemokine receptors (CXCR), such as CXCR2 and CXCR4 [56][1]. They are considered the main players in the innate immune response; however, despite their extensive studied contribution to acute liver injury, little is known about their role in NAFLD [57][2].
In this regard, several in vivo experimental studies were carried out. Intraperitoneal administration of a neutrophil-neutralizing antibody (anti-Ly6G) in a NASH-murine model (10-week HFD-fed C57BL/6 mice) reduced neutrophil liver infiltration, ameliorated metabolic features (reduction of fasting glycemia, hepatic TG content and transaminase activity), decreased hepatic inflammation (abrogation of macrophage infiltration and expression of proinflammatory cytokines such as TNFα, IL-6 and MCP-1) and profibrotic environment (reduction of profibrotic cytokine hepatic expression, such as TGF-β and α-smooth muscle actin (α-SMA)) [58][3]. Interestingly, the role of neutrophils in this context seemed to partially rely on neutrophil elastase, given that its deficiency (using a knockout murine model) improved the lipid profile and reduced hepatic damage (reduced transaminase activity, steatosis and NAS) as well as liver inflammation (decreased macrophage infiltration, TNFα and MCP-1 expression) [59][4]. Unlike the results found by Ou et al. [58][3], interleukin (IL)-6 expression was not significantly affected [59][4]. These discrepancies could account for the differential NASH induction or the involvement of other neutrophil-related mediators beyond elastase, such as myeloperoxidase (MPO). Indeed, both TNFα and IL-6 expression were downregulated in an MPO-deficient NASH-murine model, and this anti-inflammatory response was accompanied by a decrease in both neutrophil and lymphocyte hepatic infiltration, as well as by an improvement in NASH-related features, such as liver cholesterol content and the degree of fibrosis [60][5]. Accordingly, both MPO-deficiency and MPO pharmacological inhibition reduced liver damage, steatosis and fibrosis, as well as plasma levels of ALT, in a murine model of NASH, which could be explained by the existence of an MPO-dependent pathway for inflammation and apoptosis in this metabolic disease [61][6]. Moreover, there is additional evidence of MPO contribution to fibrosis development. MPO appeared to activate HSCs and to upregulate the profibrotic mediators TGF-β and α-SMA, crucial events for collagen production (Figure 1) [62][7]. In humans, plasma levels of MPO were found to be increased in NASH subjects, compared to healthy volunteers [61,63][6][8] or to patients with simple steatosis [64][9]. Furthermore, MPO mRNA hepatic expression was positively correlated with risk factors for NASH progression, such as body mass index (BMI) and the percentage of glycated hemoglobin (Table 1) [61][6].
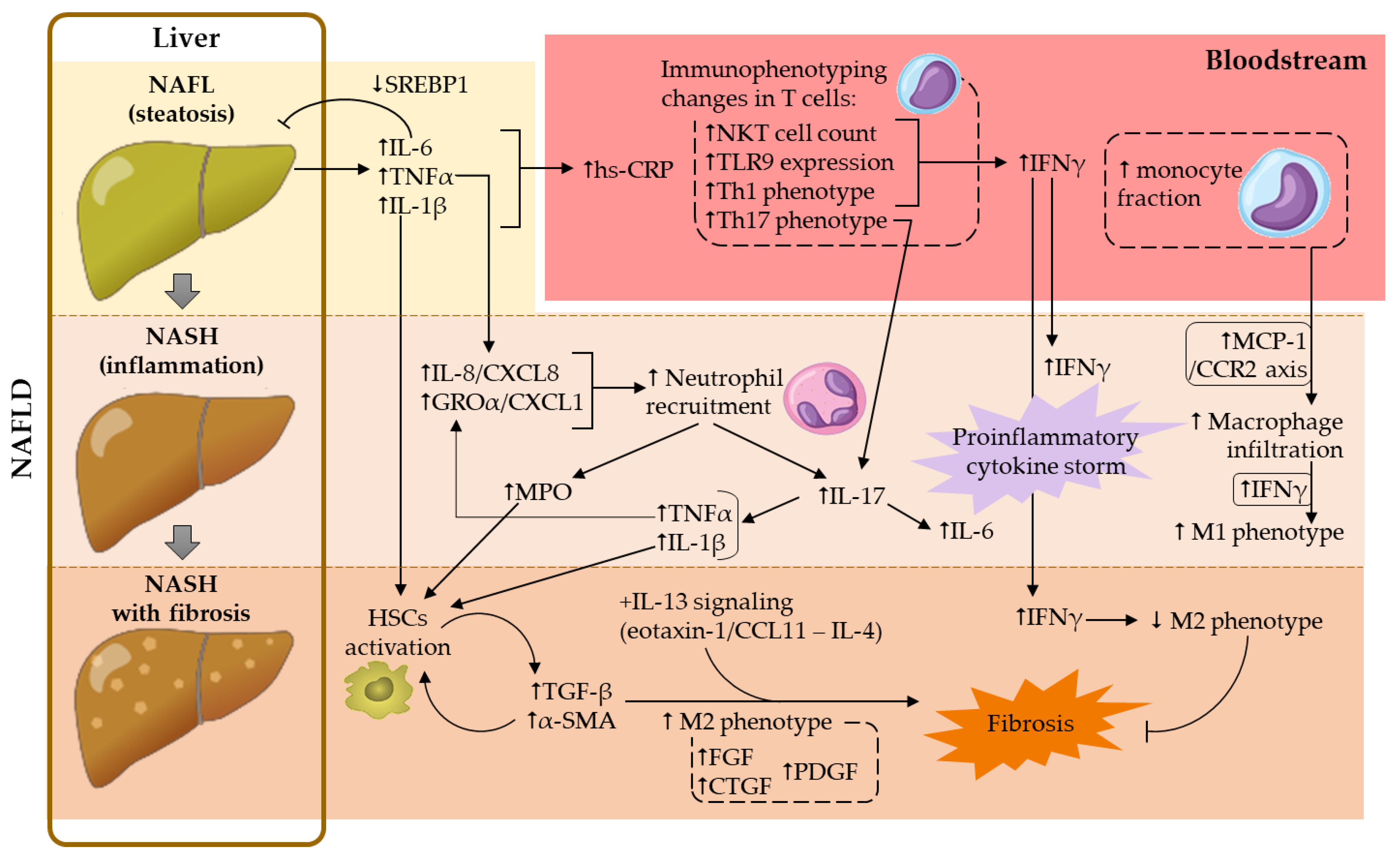
Figure 1. Contribution of the main cellular and soluble mediators studied in NAFLD development. α-SMA, α-smooth muscle actin; CTGF, connective tissue growth factor; FGF, fibroblast growth factor; GROα, growth regulated protein-α; hs-CRP, high-sensitivity C-reactive protein; HSCs, hepatic stellate cells; IFNγ, interferon-γ; IL, interleukin; MCP-1, monocyte chemoattractant protein-1; MPO, myeloperoxidase; NAFL, non-alcoholic fatty liver; NAFLD, non-alcoholic fatty liver disease; NASH, non-alcoholic steatohepatitis; NKT, natural killer T; PDGF, platelet-derived growth factor; SREBP1, sterol regulatory element binding protein-1; TGF-β, transforming growth factor-β; Th, T helper; TLR9, toll-like receptor 9; TNFα, tumor necrosis factor-α.
Table 1.
Correlations between NAFLD features and cellular or soluble markers.
| NAFLD Features | Correlation | Marker | References |
|---|---|---|---|
| Liver inflammation | + | MPV | [48][10] |
| + | Intrahepatic T-lymphocyte frequency and aggregates | [77][11] | |
| + | Circulating IL-18 levels | [142][12] | |
| + | Hepatic IL-6 expression | [150][13] | |
| NR | Peripheral percentage of IFNγ-producing T cells | [83][14] | |
| NR | Circulating MCP-1/CCL2 levels | [133][15] | |
| Steatosis | + | MPV | [48,49][10][16] |
| + | Circulating hs-CRP levels | [101,104,106][17][18][19] | |
| + | Hepatic IL-17A mRNA expression | [153][20] | |
| + | Hepatic MCP-1/CCL2 mRNA expression | [128,129,130][21][22][23] | |
| − | Peripheral activated (NKG2D+) NKT cell frequency | [20][24] | |
| NR | Peripheral percentage of IFNγ-producing T cells | [83][14] | |
| NR | Circulating MCP-1/CCL2 levels | [133][15] | |
| Fibrosis | + | MPV | [48][10] |
| + | Neutrophil/lymphocyte ratio (NLR) | [48,57,63,2][8]66,[10]67][[25][26] | |
| + | Intrahepatic T-lymphocyte frequency and aggregates | [77][11] | |
| + | Circulating IFNγ levels | [77][11] | |
| + | Circulating MCP-1/CCL2 levels | [133][15] | |
| + | Circulating IL-18 levels | [143][27] | |
| + | Circulating hs-CRP levels | [101,105][17][28] | |
| + | Hepatic IL-6 expression | [150][13] | |
| − | Hepatic NK cell frequency | [20][24] | |
| NR | Peripheral T-lymphocyte counts | [20][24] | |
| NR | Peripheral percentage of IFNγ-producing T cells | [83][14] | |
| Liver injury | + | MPV | [48][10] |
| + | TLR9 expression on circulating CD4+ or CD8+ cells | [78][29] | |
| + | Circulating MCP-1/CCL2 levels | [133][15] | |
| + | Circulating IL-18 levels | [142][12] | |
| NR | Peripheral percentage of IFNγ-producing T cells | [83][14] | |
| Body mass index | + | Hepatic MPO mRNA expression | [61][6] |
| + | TLR9 expression on circulating CD4+ or CD8+ cells | [78][29] | |
| Dysglycemia | + | Hepatic MPO mRNA expression | [61][6] |
| + | Hepatic IL-6 expression | [150][13] | |
| Hypertriglyceridemia | + | TLR9 expression on circulating CD8+ cells | [78][29] |
(+) positive correlation; (−) negative correlation; (NR) not related. Abbreviations: hs-CRP, high-sensitivity C-reactive protein; IFNγ, interferon-γ; MCP-1, monocyte chemoattractant protein-1; MPO, myeloperoxidase; MPV, mean platelet volume; TLR9, toll-like receptor 9.
On the other hand, neutrophil extracellular traps (NETs), a matrix involved in pathogen capture and destruction, appear to play a role in the early stages of NAFLD, even before monocyte-derived macrophage infiltration [65][30]. In particular, the dissolution of NETs resulted in mice protection from liver inflammation (reduction of TNFα and IL-6 expression and macrophage infiltration) and damage (diminished ALT levels), which ultimately contributed to NAS reduction [65][30].
Regarding some cell adhesion molecules (CAMs) expressed on the neutrophil surface, L-selectin (also known as CD62L, which is involved in the initial rolling) was found to be overexpressed on neutrophils from NASH patients, compared to those found in healthy controls or patients with simple steatosis. Despite these findings, no differences were observed in the neutrophilic expression of CD11b, an integrin involved in leukocyte adhesion and transmigration through the endothelium [63][8].
Furthermore, it is known that metabolic abnormalities lead to immune imbalances in peripheral blood and liver. They are manifested at the cellular level by an increased ratio of T helper (Th)17 lymphocytes to T regulatory (Treg) cells (ratio Th17/Treg) and by the dominance of neutrophils over lymphocytes. Therefore, one of the most studied parameters in liver diseases is the neutrophil/lymphocyte ratio (NLR) [66][25]. According to most of the current literature, elevated NLR values have been associated with greater severity of the disease, the evolution of liver fibrosis and the prediction of mortality in NAFLD [48,57,63,66,67][2][8][10][25][26]. However, Kara et al. found no association between NLR and the severity of liver inflammation or fibrosis in patients with NAFLD [68][31]. Nevertheless, NLR values were found to be higher in NASH patients than in healthy controls, or even in subjects affected by hepatitis B or C [56,67][1][26]. Additionally, it seems that NLR is a better predictive marker than C-reactive protein (CRP) for active chronic liver disease, and can be considered as an independent variable for predicting the occurrence of necroinflammation and fibrosis in NASH [67][26].
Along with imbalances in NLR, an altered Th17/Treg ratio also contributes to the upregulation of the above-mentioned proinflammatory (e.g., IL-6, TNFα) and profibrotic (e.g., TGF-β) cytokines, which in turn leads to a hyperactivation of the IL-17 axis, implicated in the progression of NAFL to NASH [66][25]. Indeed, it is known that hepatic human neutrophils, especially in patients with advanced NAFLD, are a relevant source of IL-17 [57,69][2][32]. IL-17 plays an important role in granulopoiesis and participates in the recruitment and infiltration of neutrophils in the initial organ injury. IL-17 also induces the production of neutrophilic cytokines and chemokines, amplifying the neutrophilic response, which aggravates the lesion (Figure 1) [66][25]. Accordingly, neutrophil depletion reduced liver inflammation and further complications in the context of NASH in mice [58][3]; however, these results could have been biased, given that these cells constitute the front line of host defense against infections.
Altogether, neutrophils appear to play a relevant role in both NAFL and NASH progression. In these diseases, neutrophil activation, increased CAMs expression on their surface, and generation and release of neutrophil-related cytokines, chemokines, enzymes and other intracellular products were detected.
2. Monocytes
Monocytes are highly plastic leukocytes that play a crucial role in host defense and tissue homeostasis, especially in bacterial and fungal elimination through phagocytic, oxidative and cytokine-producing responses [70][33]. This leukocyte subset can be divided into three functionally distinct phenotypes based on the differential expression of the surface markers CD14, CD16 and CCR2 [52,70,71][33][34][35]:
-
classical monocytes (CD14++CD16−CCR2+, also known as Mon1 subtype monocytes), represent approximately 85% of monocytes in peripheral blood. They possess a high phagocytic capacity and proinflammatory properties;
-
intermediate monocytes (CD14++CD16+CCR2+, also known as Mon2 subtype monocytes), which constitute around 5% of the total monocytes in peripheral blood;
-
nonclassical monocytes (CD14+CD16+CCR2−, also known as Mon3 subtype monocytes), represent 10% of the total monocytes in peripheral blood.
Different studies demonstrated that the total circulating leukocyte counts were elevated in patients with NAFLD, as compared to control subjects, partly due to an increase in the monocyte fraction [72,73][36][37]. Interestingly, the percentage of the Mon1 subset was found to be lower in NAFLD patients and, while Zhang et al. described an increase in the Mon2 fraction, Wang et al. documented a higher Mon3 fraction in patients with NAFLD [73,74][37][38]. Compiled evidence suggested a link between adiposity, inflammation and intermediate/nonclassical monocytes (CD16+). However, it remains unclear whether the unbalance in monocyte subtypes is a consequence of—or a contributor to—this inflammatory response. Nevertheless, monocyte fraction (along with BMI, waist circumference and plasma levels of TNFα) turned out to be an independent risk factor for NAFLD, and could become a potential prognostic biomarker in this context [73,74][37][38].
In agreement with these observations, CCR2 expression in monocytes was significantly lower in NAFLD patients, compared to control subjects [70][33], which was perhaps the consequence of imbalances between Mon1 and Mon3 fractions, as previously described [73][37]. In this regard, the development of novel drugs has shown promise. For instance, Cenicriviroc, a dual antagonist of CCR2 and CCR5 receptors, yielded satisfactory results in the first year of its phase II clinical trial, showing a significant reduction in systemic inflammation and markers of inflammation in NASH [75][39]. TLR6 was also found to be overexpressed in the monocytes of patients with NAFLD, compared to obese subjects with normal liver biopsies [76][40]. TLR6 is associated with PAMPs recognition, crucial for innate immunity activation against infectious agents. Along this line, since liver blood flow comes directly from the intestinal portal circulation, intimately linking the gut and liver, the gut microbiota profile could influence liver histology through TLR6 activation. Indeed, TLR6 deregulation found in NAFL and NASH patients seems to contribute to the worsening of liver inflammation, and has been pointed out as a potential peripheral biomarker of NASH severity [76][40].
Regarding cardiovascular complications, different studies have shown a higher incidence of subclinical atherosclerosis in patients with NAFL/NASH compared to controls (reviewed in [19][41]). Monocytes play a central role in atherosclerosis [52][34] and could play a relevant role in the development of cardiovascular diseases in NAFLD [72,73][36][37]. A possible molecular player connecting these pathologies is L-selectin/CD62L, involved in leukocyte-endothelium interactions. Its expression has been found to be increased in monocytes of patients with decompensated cirrhosis [70][33].
In summary, high monocyte counts due to a greater intermediate/nonclassical monocyte fraction appear to contribute to the development of NAFLD, its progression toward NASH and/or the development of cardiovascular complications.
3. Lymphocytes
Lymphocytes are certainly the most relevant leukocytes of the adaptive immune system. According to their activity, they are classified as T-lymphocytes (CD3+ cells), responsible for the cell-mediated responses, or B-lymphocytes (CD19+ cells), which participate in humoral/antibody responses. In turn, according to their physiological functions, T-lymphocytes are divided into different subtypes: cytotoxic T-lymphocytes (Tc or CD8+ cells) and different subsets of T-helper cells (Th or CD4+ cells), including Th1, Th2, Th17 and T regulatory lymphocytes (Treg), which seem to be involved in the pathogenesis of NAFLD [32][42]. T-helper cells are the main regulators of immune processes, orchestrating the effector functions of B-lymphocytes, cytotoxic T-lymphocytes and phagocytes. In addition, other different T-lymphocyte subtypes are associated with the innate immune system, including natural killer T-lymphocytes (NKT cells), γδ T-lymphocytes and mucosal-associated invariant T cells (MAIT cells) [20,32][24][42].
In recent years, the complex bidirectional interaction between T-lymphocytes and neutrophils has become evident, with neutrophils playing an important role in the modulation of T-lymphocyte immunological response. Antonucci et al. observed that neutrophils from NASH patients were able (ex vivo) to suppress the proliferation and activation of autologous CD4+ and CD8+ T-lymphocytes more than neutrophils from healthy donors or those with NAFL [63][8]. Potent suppression of CD4+ and CD8+ T-lymphocyte proliferation and activation could, over time, induce inadequate immune surveillance of hepatic damage, making patients more susceptible to NAFLD progression.
Positive correlations between lymphocyte aggregates (rich in T-lymphocytes), in number, size, lobular inflammation score or fibrosis staging, have been described previously (Table 1), suggesting the involvement of lymphocytes in the progression of NAFL toward NASH [77][11]. Accordingly, fibrosis staging, measured by fibroscan, and intrahepatic T-lymphocyte frequency were also found to be positively correlated. In contrast, no correlation was found between fibrosis and circulating T-lymphocyte counts (Table 1) [20][24], likely due to similar peripheral blood lymphocyte fraction or counts in NAFL and NASH compared to controls [63,72,73][8][36][37]. Nevertheless, positive correlations were found between toll-like receptor 9 (TLR9) expression in peripheral CD4+ and CD8+ T cells and clinical and pathological alterations of NAFLD (Table 1) [78][29]. Interestingly, in the same study, a downregulation of TLR9 was observed in peripheral T cells (both CD4+ and CD8+ cells) and in intrahepatic CD4+ cells of patients with NAFL, compared to control subjects. Of note, TLR9 is involved in interferon-γ (IFNγ) production by T-lymphocytes, a contributing cytokine to liver injury and inflammation (reviewed in Section 5.3). This finding may suggest a protective adaptation to hepatocellular injury in patients with simple steatosis. On the other hand, a recovery of TLR9 expression was observed in patients with NASH [78][29]. Given the complex role of IFNγ (see Section 5.3), this observation could be the consequence of a failure of this protective mechanism, contributing to a proinflammatory milieu, or a protective mechanism against liver fibrosis in these patients (Figure 1).
In addition to TLR9, other TLRs, such as TLR2, TLR4 and TLR5 have proven roles in the pathogenesis of NAFLD and its progression to NASH (reviewed in [79][43]). For example, TLR4, mRNA is overexpressed in the liver of patients with NASH compared to patients with NAFL [80][44], and TLR4 deficiency in ob/ob mice protects the liver from damage and hepatitis, but not from steatosis [81][45]. Although TLRs are important IFNγ-regulating factors, the generation and release of this proinflammatory cytokine is also regulated by other mediators, such as substances secreted by different enteric bacteria in NAFLD [79][43].
Thus, more comprehensive studies should be performed to better understand the role of the different lymphocyte subpopulations in both NAFL and NASH. The main findings regarding the different lymphocyte subsets in this field are described below.
3.1. Th1 Cells
Th1 lymphocytes are proinflammatory cells characterized by the production of IFNγ, IL-2 and TNFα. The main cytokines involved in the differentiation of Th0 (naïve T cell) toward the Th1 phenotype are IL-12 and IFNγ, through the activation of signal transducers and activators of transcription (STAT) 1 and STAT4. These cells play an important role in the cellular component of the adaptive immune system (cell-mediated immunity), especially in the host defense against intracellular pathogens, through macrophage activation [32][42].
Regarding Th1 frequency, there is evidence of a greater peripheral percentage of IFNγ-producing CD4+ cells in NAFL and NASH than in control subjects [78,82,83,84,85][14][29][46][47][48]. Despite these findings, no significant correlations were detected between the peripheral percentage of IFNγ-producing CD4+ cells and histological features of NASH, such as steatosis, lobular inflammation, ballooning and fibrosis stage [83][14].
Regarding intrahepatic examination, Rau et al. documented higher percentages of IFNγ-producing CD4+ cells in the liver, compared to circulating values, in both NAFL and NASH [82][46]. These results could indicate an enhanced liver infiltration of Th1 cells in these hepatic complications. Nevertheless, determinations in control subjects are required in order to provide proof. Liver upregulation of genes associated with the promotion of Th1 phenotype was described in NASH patients compared to those with NAFLD or obese patients [86][49].
Despite the above-mentioned results, human studies regarding Th1 cells in NAFLD remain, to date, insufficient sources from which to draw accurate conclusions.
3.2. Th2 Cells
Th2 cell differentiation from naïve T cells is mainly driven by IL-2 and IL-4. Functionally, Th2 lymphocytes are involved in host defenses against parasitic infections and play a prominent role in the pathogenesis of allergic diseases. These cells produce several cytokines, including IL-4, IL-5, IL-10 and IL-13, most of them relevant to the orchestration of humoral immunity, through the activation of STAT5 and STAT6 [32][42].
Very few human studies have investigated the role of this lymphocyte subtype in both NAFL and NASH, and the findings of these remain controversial. While some studies did not find differences in the percentage of IL-4-producing CD4+ cells (considered as Th2 cells) in the peripheral blood of NASH patients compared to control subjects [83[14][48],85], other studies found a greater percentage of these circulating cells in both NAFL and NASH patients compared to healthy volunteers [20,82][24][46]. Although no differences were observed between NAFL and NASH patients, Rau et al. documented a higher circulating Th2/Treg ratio in patients with NASH compared to those with NAFLD, which was significantly reduced 1 year after bariatric surgery in these NASH patients [82][46].
Regarding the Th2 cell frequency in liver tissue, a greater percentage of IL-4-producing CD4+ cells was described, compared to circulating values in both NAFL and NASH patients [73][37]. Although this observation could indicate an increased liver infiltration of Th2 cells, existing data from control subjects are insufficient to prove it.
Given the increased circulating Th2 cell counts in NAFLD patients documented by some authors, along with the apparent enhanced Th2 cell liver infiltration, it seems that this T cell subset likely plays a yet unknown role in NAFLD development and/or progression to NASH.
3.3. Th17 Cells
The differentiation of naïve T cells into Th17 is orchestrated by several cytokines, including TGF-β, IL-1β, IL-6, IL-21, IL-23 and TNFα, through the activation of STAT3. These proinflammatory cells are relevant in cell-mediated immunity, especially for the host defense against extracellular pathogens, through the generation and release of proinflammatory cytokines such as IL-17 (mainly the isoforms IL-17A and IL-17F), IL-22 and IL-23 [32][42]. These cytokines are responsible for the synthesis of some neutrophil chemoattractant chemokines, such as GROα/CXCL1, GROβ/CXCL2 or IL-8/CXCL8 [87][50].
In the context of NAFLD, IL-17 seems to be the most relevant Th17-related cytokine. IL-17 exacerbates liver inflammation by increasing leukocyte infiltration, stimulating the generation of other proinflammatory mediators and promoting profibrotic effects (reviewed in [66][25]). The strong proinflammatory response induced by IL-17 is due to the ubiquitous expression of its counterreceptor (IL-17r), which is localized on endothelial and epithelial cells, as well as on monocytes and macrophages [32][42].
Th17 cells have been studied more extensively in these hepatic complications, but again, findings are not exempt from discrepancies. Whereas Wang et al. described a greater percentage of IL-17-producing CD4+ cells (considered as Th17 cells) in the peripheral blood of NASH patients compared to NAFL or healthy controls [84][47], Rau et al. did not find any differences [82][46]. These dissimilarities could have been reliant on the differential diagnostic approaches employed. NAFL and NASH were diagnosed accurately via liver biopsy and liver functional tests by Wang et al. [84][47], while a noninvasive sonographic NASH score was used by Rau et al. [82][46]. Nevertheless, the latter study reported a higher circulating Th17/Treg ratio in NASH patients compared to NAFL or healthy controls, which was significantly reduced 1 year after bariatric surgery of NASH patients [82][46].
Moreover, a greater percentage of hepatic IL-17-producing CD4+ cells was found in both NAFL and NASH patients compared to circulating values [73][37]. In agreement with this observation, Tang et al. reported a greater IL-17+ cell infiltration in liver tissue of NASH patients compared to control subjects, using immunohistochemical analyses, and also described increased liver mRNA expression of IL-17 and other Th17-related cytokines, such as IL-21 and IL-23 [88][51]. Liver biopsies from NASH patients presented a higher percentage of Th17 cells and Th17/Treg ratios than those from NAFL subjects [82][46]. Taken together, these results suggested that Th17 cells, through IL-17 signaling, could be crucial for NAFL progression to NASH.
In addition, circulating levels of IL-17 were found to be increased in NASH patients compared to control subjects. Interestingly, IL-17 levels were significantly higher in those NASH patients with fibrosis than in non-fibrotic patients [89][52]. These results emphasized the potential link between the IL-17 axis and TGF-β signaling in NASH [66][25].
Therefore, Th17 cells, through the activation of the IL-17 axis, could play a pivotal role in this liver disorder, especially in the progression of NAFL to NASH and its further complications.
3.4. Treg Cells
According to their origin, circulating Treg cells can be divided into two main subsets: those matured in the thymus (tTreg) and those differentiated from naïve T cells, mainly induced by the action of TGF-β (iTreg). Both subsets exert immunoregulatory roles on Th1 and Th17 responses generating anti-inflammatory effects. Treg cells produce important cytokines for immunoregulation, including TGF-β, which amplifies the Treg differentiation from naïve T cells, and the anti-inflammatory IL-10 [32][42]. However, it is important to highlight that TGF-β, along with IL-6, is also responsible for Th17 cell differentiation from T naïve cells, and possesses profibrotic properties, reflecting the complexity of Th17/Treg balance [32][42].
Regarding the frequency and role of Treg cells in NAFLD, very few human studies are currently available. According to the scientific literature, and as mentioned above, unbalances in Th1/Treg, Th2/Treg and Th17/Treg ratios were observed in these hepatic complications [82][46]. In particular, at the circulating level, while both Th1/Treg and Th2/Treg ratios were significantly higher in NAFL or NASH patients than in control subjects, Th17/Treg ratio was only greater in NASH patients, as compared to both NAFL and control subjects. Within the liver, only the Th17/Treg ratio was higher in NASH patients, compared to NAFL patients [82][46]. Of note, the percentage of circulating CD25++CD4+ cells (considered by the authors as Treg cells) was significantly higher in NASH patients 1 year after bariatric surgery, which was accompanied by a reduction in both circulating Th2/Treg and Th17/Treg ratios [82][46].
Likewise, a decreased circulating IL-10/IL-17 ratio was detected in NASH patients, compared to non-NASH obese patients [90][53]. Overall, these findings suggested a link between NAFL development and its progression to NASH with an unfavorable Treg cell balance (decreased Treg cell fraction and/or increased Th1/2/17 frequencies).
3.5. CD8+ T Cells
CD8+ T lymphocytes, commonly known as cytotoxic T cells, are formed in the bone marrow and matured in the thymus. They are pivotal players in the elimination of infected or tumoral cells, through the recognition of antigens presented by the major histocompatibility complex (MHC) class I. These cytotoxic effects are achieved through the secretion of cytokines (e.g., IFNγ), cytotoxic agents (e.g., perforin and granzyme) and direct cell-cell contact [32][42].
Given their effects against tumoral cells, the role of CD8+ cells in NASH-related liver cancer has been extensively studied [91,92][54][55]. However, their role in the development of NAFLD and its progression to non-cancerous NASH is limited. According to the current literature, NASH patients and control individuals present similar circulating CD8+ cell counts [82][46]. However, a greater percentage of IFNγ-producing CD8+ cells was observed in the peripheral blood of NASH patients, compared to control subjects [78,82,85][29][46][48].
As previously outlined, positive correlations were found between the expression of TLR9 in circulating CD8+ T cells and clinical features of NAFLD, such as BMI, TG levels and liver transaminase levels [78][29]. The observed downregulation of TLR9 in CD8+ cells of patients with NAFL, compared to control subjects, could imply a protective response to hepatocellular injury, since TLR9 is involved in IFNγ production by CD8+ cells [78][29]. However, no differences were found between NASH patients and control subjects [78][29], possibly due to a failure in this protective mechanism (contributing to a proinflammatory milieu) or to a protective mechanism against liver fibrosis in these patients. In agreement with these observations, Inzaugarat et al. found no significant correlations between the increased percentages of IFNγ-producing CD8+ cells and histological features of NASH (Table 1) [83][14].
In contrast, the expression of CD69 (a well-known marker of early lymphocyte activation) was found to be upregulated in peripheral CD8+ cells of patients with NASH, compared to NAFL subjects, suggesting that CD8+ cell activation may be involved in the progression of NAFL to NASH [78][29]. Despite these findings, little is known regarding the liver infiltration of CD8+ cells in these metabolic disorders in humans and further studies are required.
3.6. Natural Killer (NK) and Natural Killer T (NKT) Cells
In addition to T and B cells, there is another group of lymphocytes, the so-called natural killer (NK) cells. Given their crucial role in the elimination of aberrant cells, they are considered relevant cellular players in the innate immune system. NK cell-related cytotoxic activity is due to the release of cytotoxic mediators (e.g., granzyme and perforin) from their granules, as well as the generation of IFNγ, which enhances both NK and CD8+ cell activities against tumor cells [93][56].
In addition to their role in the immune surveillance of abnormal cells, the role of these cells on NAFL or NASH pathogenesis has been addressed in humans [20,93,94][24][56][57]. Nevertheless, inconsistent results were obtained, likely due to the complexity of liver disease and heterogeneity among NK cells. While some studies described a reduced circulating NK cell (both CD56bright and CD56dim subsets) frequency in NAFLD patients, compared to control subjects [20[24][56],93], Stiglund et al. did not find any significant differences [94][57]. Additionally, the augmented frequency of highly dysfunctional Siglec7−CD57+PD-1+CD56dim NK cell subsets in NAFLD patient bloodstreams demonstrated a functional impairment of NK cells in this context [93][56]. These studies analyzed liver biopsies for NAFLD diagnosis. However, the observed discrepancies were likely derived from differences in NAFLD patients’ stratification. While Diedrich et al. only compared NAFLD patients with control subjects [20][24], Sakamoto et al. analyzed results between patients with no or mild fibrosis vs. patients with advanced fibrosis [93][56], and Stiglund et al. analyzed 3 groups: NASH patients vs. NAFL patients vs. control subjects [94][57].
In the analysis of liver biopsies, a greater percentage of CD56dim NK cells was observed in NAFLD patients, which could account for their decreased percentage in circulation [20][24]. Nonetheless, the same study revealed a negative correlation between total NK cell frequency in the liver and the fibrosis stage measured by fibroscan elastography (Table 1) [20][24].
NKG2D and CD69 are markers of NK cell activation. In this context, while some studies documented higher peripheral and liver NK cell activation in NAFL/NASH patients [93[56][57],94], Diedrich et al. described the opposite in circulation [20][24].
On the other hand, there is a CD3+ T-lymphocyte subset (CD3 is not expressed by NK cells) that expresses some NK cell markers (CD56 or CD161), the so-called NKT cells. In NAFLD, a greater proportion of NKT cells in both blood and liver was detected, which seemed to be associated with the severity of the disease [95,96][58][59]. Again, there have been few (and contradictory) results regarding NKT cell activation. Indeed, both greater and reduced circulating NKT cell activation have been associated with this complex disease [20,97][24][60]. The frequency of circulating NKG2D+ NKT cells was negatively correlated with the grade of steatosis (Table 1) [20][24].
Similarly, although several attempts to investigate the role of NK/NKT cells in NAFLD have been carried out, their role in this disease has remained elusive.
References
- Wu, L.; Gao, X.; Guo, Q.; Li, J.; Yao, J.; Yan, K.; Xu, Y.; Jiang, X.; Ye, D.; Guo, J. The role of neutrophils in innate immunity-driven nonalcoholic steatohepatitis: Lessons learned and future promise. Hepatol. Int. 2020, 14, 652–666.
- Nati, M.; Chung, K.J.; Chavakis, T. The Role of Innate Immune Cells in Nonalcoholic Fatty Liver Disease. J. Innate Immun. 2022, 14, 31–41.
- Ou, R.; Liu, J.; Lv, M.; Wang, J.; Wang, J.; Zhu, L.; Zhao, L.; Xu, Y. Neutrophil depletion improves diet-induced non-alcoholic fatty liver disease in mice. Endocrine 2017, 57, 72–82.
- Chen, J.; Liang, B.; Bian, D.; Luo, Y.; Yang, J.; Li, Z.; Zhuang, Z.; Zang, S.; Shi, J. Knockout of neutrophil elastase protects against western diet induced nonalcoholic steatohepatitis in mice by regulating hepatic ceramides metabolism. Biochem. Biophys. Res. Commun. 2019, 518, 691–697.
- Rensen, S.S.; Bieghs, V.; Xanthoulea, S.; Arfianti, E.; Bakker, J.A.; Shiri-Sverdlov, R.; Hofker, M.H.; Greve, J.W.; Buurman, W.A. Neutrophil-derived myeloperoxidase aggravates non-alcoholic steatohepatitis in low-density lipoprotein receptor-deficient mice. PLoS ONE 2012, 7, e52411.
- Koop, A.C.; Thiele, N.D.; Steins, D.; Michaëlsson, E.; Wehmeyer, M.; Scheja, L.; Steglich, B.; Huber, S.; Schulze Zur Wiesch, J.; Lohse, A.W.; et al. Therapeutic Targeting of Myeloperoxidase Attenuates NASH in Mice. Hepatol. Commun. 2020, 4, 1441–1458.
- Pulli, B.; Ali, M.; Iwamoto, Y.; Zeller, M.W.; Schob, S.; Linnoila, J.J.; Chen, J.W. Myeloperoxidase-Hepatocyte-Stellate Cell Cross Talk Promotes Hepatocyte Injury and Fibrosis in Experimental Nonalcoholic Steatohepatitis. Antioxid. Redox Signal. 2015, 23, 1255–1269.
- Antonucci, L.; Porcu, C.; Timperi, E.; Santini, S.J.; Iannucci, G.; Balsano, C. Circulating Neutrophils of Nonalcoholic Steatohepatitis Patients Show an Activated Phenotype and Suppress T Lymphocytes Activity. J. Immunol. Res. 2020, 2020, 4570219.
- Rensen, S.S.; Slaats, Y.; Nijhuis, J.; Jans, A.; Bieghs, V.; Driessen, A.; Malle, E.; Greve, J.W.; Buurman, W.A. Increased hepatic myeloperoxidase activity in obese subjects with nonalcoholic steatohepatitis. Am. J. Pathol. 2009, 175, 1473–1482.
- Abdel-Razik, A.; Mousa, N.; Shabana, W.; Refaey, M.; ElMahdy, Y.; Elhelaly, R.; Elzehery, R.; Zalata, K.; Arafa, M.; Elbaz, S.; et al. A novel model using mean platelet volume and neutrophil to lymphocyte ratio as a marker of nonalcoholic steatohepatitis in NAFLD patients: Multicentric study. Eur. J. Gastroenterol. Hepatol. 2016, 28, e1–e9.
- Bruzzì, S.; Sutti, S.; Giudici, G.; Burlone, M.E.; Ramavath, N.N.; Toscani, A.; Bozzola, C.; Schneider, P.; Morello, E.; Parola, M.; et al. B2-Lymphocyte responses to oxidative stress-derived antigens contribute to the evolution of nonalcoholic fatty liver disease (NAFLD). Free Radic. Biol. Med. 2018, 124, 249–259.
- Ludwiczek, O.; Kaser, A.; Novick, D.; Dinarello, C.A.; Rubinstein, M.; Vogel, W.; Tilg, H. Plasma levels of interleukin-18 and interleukin-18 binding protein are elevated in patients with chronic liver disease. J. Clin. Immunol. 2002, 22, 331–337.
- Wieckowska, A.; Papouchado, B.G.; Li, Z.; Lopez, R.; Zein, N.N.; Feldstein, A.E. Increased hepatic and circulating interleukin-6 levels in human nonalcoholic steatohepatitis. Am. J. Gastroenterol. 2008, 103, 1372–1379.
- Inzaugarat, M.E.; Ferreyra Solari, N.E.; Billordo, L.A.; Abecasis, R.; Gadano, A.C.; Cherñavsky, A.C. Altered phenotype and functionality of circulating immune cells characterize adult patients with nonalcoholic steatohepatitis. J. Clin. Immunol. 2011, 31, 1120–1130.
- Puengel, T.; Lefere, S.; Hundertmark, J.; Kohlhepp, M.; Penners, C.; Van de Velde, F.; Lapauw, B.; Hoorens, A.; Devisscher, L.; Geerts, A.; et al. Combined Therapy with a CCR2/CCR5 Antagonist and FGF21 Analogue Synergizes in Ameliorating Steatohepatitis and Fibrosis. Int. J. Mol. Sci. 2022, 23, 6696.
- Alkhouri, N.; Kistangari, G.; Campbell, C.; Lopez, R.; Zein, N.N.; Feldstein, A.E. Mean platelet volume as a marker of increased cardiovascular risk in patients with nonalcoholic steatohepatitis. Hepatology 2012, 55, 331.
- Duan, Y.; Pan, X.; Luo, J.; Xiao, X.; Li, J.; Bestman, P.L.; Luo, M. Association of Inflammatory Cytokines with Non-Alcoholic Fatty Liver Disease. Front. Immunol. 2022, 13, 880298.
- Kumar, R.; Porwal, Y.C.; Dev, N.; Kumar, P.; Chakravarthy, S.; Kumawat, A. Association of high-sensitivity C-reactive protein (hs-CRP) with non-alcoholic fatty liver disease (NAFLD) in Asian Indians: A cross-sectional study. J. Fam. Med. Prim. Care 2020, 9, 390–394.
- Zimmermann, E.; Anty, R.; Tordjman, J.; Verrijken, A.; Gual, P.; Tran, A.; Iannelli, A.; Gugenheim, J.; Bedossa, P.; Francque, S.; et al. C-reactive protein levels in relation to various features of non-alcoholic fatty liver disease among obese patients. J. Hepatol. 2011, 55, 660–665.
- Gomes, A.L.; Teijeiro, A.; Burén, S.; Tummala, K.S.; Yilmaz, M.; Waisman, A.; Theurillat, J.P.; Perna, C.; Djouder, N. Metabolic Inflammation-Associated IL-17A Causes Non-alcoholic Steatohepatitis and Hepatocellular Carcinoma. Cancer Cell 2016, 30, 161–175.
- Greco, D.; Kotronen, A.; Westerbacka, J.; Puig, O.; Arkkila, P.; Kiviluoto, T.; Laitinen, S.; Kolak, M.; Fisher, R.M.; Hamsten, A.; et al. Gene expression in human NAFLD. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, G1281–G1287.
- Westerbacka, J.; Kolak, M.; Kiviluoto, T.; Arkkila, P.; Sirén, J.; Hamsten, A.; Fisher, R.M.; Yki-Järvinen, H. Genes involved in fatty acid partitioning and binding, lipolysis, monocyte/macrophage recruitment, and inflammation are overexpressed in the human fatty liver of insulin-resistant subjects. Diabetes 2007, 56, 2759–2765.
- Liu, Y.; Liu, X.; Zhou, W.; Zhang, J.; Wu, J.; Guo, S.; Jia, S.; Wang, H.; Li, J.; Tan, Y. Integrated bioinformatics analysis reveals potential mechanisms associated with intestinal flora intervention in nonalcoholic fatty liver disease. Medicine 2022, 101, e30184.
- Diedrich, T.; Kummer, S.; Galante, A.; Drolz, A.; Schlicker, V.; Lohse, A.W.; Kluwe, J.; Eberhard, J.M.; Schulze zur Wiesch, J. Characterization of the immune cell landscape of patients with NAFLD. PLoS ONE 2020, 15, e0230307.
- Paquissi, F.C. Immune Imbalances in Non-Alcoholic Fatty Liver Disease: From General Biomarkers and Neutrophils to Interleukin-17 Axis Activation and New Therapeutic Targets. Front. Immunol. 2016, 7, 490.
- Yilmaz, H.; Yalcin, K.S.; Namuslu, M.; Celik, H.T.; Sozen, M.; Inan, O.; Nadir, I.; Turkay, C.; Akcay, A.; Kosar, A. Neutrophil-Lymphocyte Ratio (NLR) Could Be Better Predictor than C-reactive Protein (CRP) for Liver Fibrosis in Non-alcoholic Steatohepatitis(NASH). Ann. Clin. Lab. Sci. 2015, 45, 278–286.
- Mehta, R.; Neupane, A.; Wang, L.; Goodman, Z.; Baranova, A.; Younossi, Z.M. Expression of NALPs in adipose and the fibrotic progression of non-alcoholic fatty liver disease in obese subjects. BMC Gastroenterol. 2014, 14, 208.
- Yoneda, M.; Mawatari, H.; Fujita, K.; Iida, H.; Yonemitsu, K.; Kato, S.; Takahashi, H.; Kirikoshi, H.; Inamori, M.; Nozaki, Y.; et al. High-sensitivity C-reactive protein is an independent clinical feature of nonalcoholic steatohepatitis (NASH) and also of the severity of fibrosis in NASH. J. Gastroenterol. 2007, 42, 573–582.
- Alegre, N.S.; Garcia, C.C.; Billordo, L.A.; Ameigeiras, B.; Poncino, D.; Benavides, J.; Colombato, L.; Cherñavsky, A.C. Limited expression of TLR9 on T cells and its functional consequences in patients with nonalcoholic fatty liver disease. Clin. Mol. Hepatol. 2020, 26, 216–226.
- van der Windt, D.J.; Sud, V.; Zhang, H.; Varley, P.R.; Goswami, J.; Yazdani, H.O.; Tohme, S.; Loughran, P.; O’Doherty, R.M.; Minervini, M.I.; et al. Neutrophil extracellular traps promote inflammation and development of hepatocellular carcinoma in nonalcoholic steatohepatitis. Hepatology 2018, 68, 1347–1360.
- Kara, M.; Dogru, T.; Genc, H.; Sertoglu, E.; Celebi, G.; Gurel, H.; Kayadibi, H.; Cicek, A.F.; Ercin, C.N.; Sonmez, A. Neutrophil-to-lymphocyte ratio is not a predictor of liver histology in patients with nonalcoholic fatty liver disease. Eur. J. Gastroenterol. Hepatol. 2015, 27, 1144–1148.
- Giles, D.A.; Moreno-Fernandez, M.E.; Divanovic, S. IL-17 Axis Driven Inflammation in Non-Alcoholic Fatty Liver Disease Progression. Curr. Drug Targets 2015, 16, 1315–1323.
- Gadd, V.L.; Patel, P.J.; Jose, S.; Horsfall, L.; Powell, E.E.; Irvine, K.M. Altered Peripheral Blood Monocyte Phenotype and Function in Chronic Liver Disease: Implications for Hepatic Recruitment and Systemic Inflammation. PLoS ONE 2016, 11, e0157771.
- Marques, P.; Collado, A.; Martinez-Hervás, S.; Domingo, E.; Benito, E.; Piqueras, L.; Real, J.T.; Ascaso, J.F.; Sanz, M.-J. Systemic Inflammation in Metabolic Syndrome: Increased Platelet and Leukocyte Activation, and Key Role of CX(3)CL1/CX(3)CR1 and CCL2/CCR2 Axes in Arterial Platelet-Proinflammatory Monocyte Adhesion. J. Clin. Med. 2019, 8, 708.
- Weber, C.; Shantsila, E.; Hristov, M.; Caligiuri, G.; Guzik, T.; Heine, G.H.; Hoefer, I.E.; Monaco, C.; Peter, K.; Rainger, E.; et al. Role and analysis of monocyte subsets in cardiovascular disease. Joint consensus document of the European Society of Cardiology (ESC) Working Groups “Atherosclerosis & Vascular Biology” and “Thrombosis”. Thromb. Haemost. 2016, 116, 626–637.
- Kim, H.L.; Chung, G.E.; Park, I.Y.; Choi, J.M.; Hwang, S.M.; Lee, J.H.; Kim, D. Elevated peripheral blood monocyte fraction in nonalcoholic fatty liver disease. Tohoku J. Exp. Med. 2011, 223, 227–233.
- Wang, Y.; Oeztuerk, S.; Kratzer, W.; Boehm, B.O. A Nonclassical Monocyte Phenotype in Peripheral Blood is Associated with Nonalcoholic Fatty Liver Disease: A Report from an EMIL Subcohort. Horm. Metab. Res. = Horm.-Und Stoffwechs. = Horm. Et Metab. 2016, 48, 54–61.
- Zhang, J.; Chen, W.; Fang, L.; Li, Q.; Zhang, X.; Zhang, H.; Guan, Q.; Zhao, R.; Yang, C.; Jing, F. Increased intermediate monocyte fraction in peripheral blood is associated with nonalcoholic fatty liver disease. Wien. Klin. Wochenschr. 2018, 130, 390–397.
- Vonghia, L.; Van Herck, M.A.; Weyler, J.; Francque, S. Targeting Myeloid-Derived Cells: New Frontiers in the Treatment of Non-alcoholic and Alcoholic Liver Disease. Front. Immunol. 2019, 10, 563.
- Arias-Loste, M.T.; Iruzubieta, P.; Puente, Á.; Ramos, D.; Santa Cruz, C.; Estébanez, Á.; Llerena, S.; Alonso-Martín, C.; San Segundo, D.; Álvarez, L.; et al. Increased Expression Profile and Functionality of TLR6 in Peripheral Blood Mononuclear Cells and Hepatocytes of Morbidly Obese Patients with Non-Alcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2016, 17, 1878.
- Tana, C.; Ballestri, S.; Ricci, F.; Di Vincenzo, A.; Ticinesi, A.; Gallina, S.; Giamberardino, M.A.; Cipollone, F.; Sutton, R.; Vettor, R.; et al. Cardiovascular Risk in Non-Alcoholic Fatty Liver Disease: Mechanisms and Therapeutic Implications. Int. J. Environ. Res. Public Health 2019, 16, 3104.
- Van Herck, M.A.; Weyler, J.; Kwanten, W.J.; Dirinck, E.L.; De Winter, B.Y.; Francque, S.M.; Vonghia, L. The Differential Roles of T Cells in Non-alcoholic Fatty Liver Disease and Obesity. Front. Immunol. 2019, 10, 82.
- Miura, K.; Ohnishi, H. Role of gut microbiota and Toll-like receptors in nonalcoholic fatty liver disease. World J. Gastroenterol. 2014, 20, 7381–7391.
- Sharifnia, T.; Antoun, J.; Verriere, T.G.; Suarez, G.; Wattacheril, J.; Wilson, K.T.; Peek, R.M., Jr.; Abumrad, N.N.; Flynn, C.R. Hepatic TLR4 signaling in obese NAFLD. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 309, G270–G278.
- Sutter, A.G.; Palanisamy, A.P.; Lench, J.H.; Jessmore, A.P.; Chavin, K.D. Development of steatohepatitis in Ob/Ob mice is dependent on Toll-like receptor 4. Ann. Hepatol. 2015, 14, 735–743.
- Rau, M.; Schilling, A.K.; Meertens, J.; Hering, I.; Weiss, J.; Jurowich, C.; Kudlich, T.; Hermanns, H.M.; Bantel, H.; Beyersdorf, N.; et al. Progression from Nonalcoholic Fatty Liver to Nonalcoholic Steatohepatitis Is Marked by a Higher Frequency of Th17 Cells in the Liver and an Increased Th17/Resting Regulatory T Cell Ratio in Peripheral Blood and in the Liver. J. Immunol. 2016, 196, 97–105.
- Wang, X.; Ji, D.; Zhu, B.; Jiang, S.; Han, L.; Wang, Y.; Mai, H.; Xu, S.; Jiang, H.; Wang, G.; et al. Contribution of endotoxin to Th17 bias in patients with non-alcoholic steatohepatitis. Microb. Pathog. 2020, 142, 104009.
- Ferreyra Solari, N.E.; Inzaugarat, M.E.; Baz, P.; De Matteo, E.; Lezama, C.; Galoppo, M.; Galoppo, C.; Cherñavsky, A.C. The role of innate cells is coupled to a Th1-polarized immune response in pediatric nonalcoholic steatohepatitis. J. Clin. Immunol. 2012, 32, 611–621.
- Bertola, A.; Bonnafous, S.; Anty, R.; Patouraux, S.; Saint-Paul, M.C.; Iannelli, A.; Gugenheim, J.; Barr, J.; Mato, J.M.; Le Marchand-Brustel, Y.; et al. Hepatic expression patterns of inflammatory and immune response genes associated with obesity and NASH in morbidly obese patients. PLoS ONE 2010, 5, e13577.
- Onishi, R.M.; Gaffen, S.L. Interleukin-17 and its target genes: Mechanisms of interleukin-17 function in disease. Immunology 2010, 129, 311–321.
- Tang, Y.; Bian, Z.; Zhao, L.; Liu, Y.; Liang, S.; Wang, Q.; Han, X.; Peng, Y.; Chen, X.; Shen, L.; et al. Interleukin-17 exacerbates hepatic steatosis and inflammation in non-alcoholic fatty liver disease. Clin. Exp. Immunol. 2011, 166, 281–290.
- Serhal, R.; Hilal, G.; Boutros, G.; Sidaoui, J.; Wardi, L.; Ezzeddine, S.; Alaaeddine, N. Nonalcoholic Steatohepatitis: Involvement of the Telomerase and Proinflammatory Mediators. Biomed Res. Int. 2015, 2015, 850246.
- Vonghia, L.; Magrone, T.; Verrijken, A.; Michielsen, P.; Van Gaal, L.; Jirillo, E.; Francque, S. Peripheral and Hepatic Vein Cytokine Levels in Correlation with Non-Alcoholic Fatty Liver Disease (NAFLD)-Related Metabolic, Histological, and Haemodynamic Features. PLoS ONE 2015, 10, e0143380.
- Mirshahi, F.; Aqbi, H.F.; Isbell, M.; Manjili, S.H.; Guo, C.; Saneshaw, M.; Bandyopadhyay, D.; Dozmorov, M.; Khosla, A.; Wack, K.; et al. Distinct hepatic immunological patterns are associated with the progression or inhibition of hepatocellular carcinoma. Cell Rep. 2022, 38, 110454.
- Pfister, D.; Núñez, N.G.; Pinyol, R.; Govaere, O.; Pinter, M.; Szydlowska, M.; Gupta, R.; Qiu, M.; Deczkowska, A.; Weiner, A.; et al. NASH limits anti-tumour surveillance in immunotherapy-treated HCC. Nature 2021, 592, 450–456.
- Sakamoto, Y.; Yoshio, S.; Doi, H.; Mori, T.; Matsuda, M.; Kawai, H.; Shimagaki, T.; Yoshikawa, S.; Aoki, Y.; Osawa, Y.; et al. Increased Frequency of Dysfunctional Siglec-7(-)CD57(+)PD-1(+) Natural Killer Cells in Patients With Non-alcoholic Fatty Liver Disease. Front. Immunol. 2021, 12, 603133.
- Stiglund, N.; Strand, K.; Cornillet, M.; Stål, P.; Thorell, A.; Zimmer, C.L.; Näslund, E.; Karlgren, S.; Nilsson, H.; Mellgren, G.; et al. Retained NK Cell Phenotype and Functionality in Non-alcoholic Fatty Liver Disease. Front. Immunol. 2019, 10, 1255.
- Adler, M.; Taylor, S.; Okebugwu, K.; Yee, H.; Fielding, C.; Fielding, G.; Poles, M. Intrahepatic natural killer T cell populations are increased in human hepatic steatosis. World J. Gastroenterol. 2011, 17, 1725–1731.
- Tajiri, K.; Shimizu, Y.; Tsuneyama, K.; Sugiyama, T. Role of liver-infiltrating CD3+CD56+ natural killer T cells in the pathogenesis of nonalcoholic fatty liver disease. Eur. J. Gastroenterol. Hepatol. 2009, 21, 673–680.
- Maricic, I.; Marrero, I.; Eguchi, A.; Nakamura, R.; Johnson, C.D.; Dasgupta, S.; Hernandez, C.D.; Nguyen, P.S.; Swafford, A.D.; Knight, R.; et al. Differential Activation of Hepatic Invariant NKT Cell Subsets Plays a Key Role in Progression of Nonalcoholic Steatohepatitis. J. Immunol. 2018, 201, 3017–3035.
More