Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Sirius Huang and Version 1 by Ka Shing Cheung.
Gut microbiota is increasingly recognized to play a pivotal role in various human physiological functions and diseases. Amidst the COVID-19 pandemic, research has suggested that dysbiosis of the gut microbiota is also involved in the development and severity of COVID-19 symptoms by regulating SARS-CoV-2 entry and modulating inflammation.
- gut microbiota
- gut dysbiosis
- COVID-19
- COVID-19 vaccine
1. Introduction
As it has been increasingly shown that gut microbiota plays a pivotal role in the human immune system, the association between the gut microbiota and COVID-19 has been extensively studied during the pandemic. A considerable number of cross-sectional studies performed on animals and humans alike demonstrated that gut microbiota dysbiosis was observed during SARS-CoV-2 infection, though whether this was the cause or the effect of SARS-CoV-2 infection remained not fully understood. Nonetheless, gut microbiota dysbiosis appears to modulate COVID-19 severity and clinical outcomes, while in turn SARS-CoV-2 infection may induce alterations in the gut microbiota. As such, gut microbiota dysbiosis is hypothesized to have a bidirectional relationship with COVID-19 and its outcomes.
2. Gut Microbiota Dysbiosis Associated with Susceptibility to SARS-CoV-2 Infection and Disease Severity
While evidence that directly implicates the gut microbiota in affecting a person’s susceptibility to SARS-CoV-2 is still currently lacking, it has been suggested that the gut microbiota dysbiosis can increase the risk of SARS-CoV-2 infection by modulating the expression of the viral entry receptor angiotensin-converting enzyme 2 (ACE2) in the gut and by regulating B cells and T cells [8][1]. Animal and human studies, however, have shown that gut microbiota dysbiosis could be associated with more severe clinical outcomes. In a study conducted on healthy hamsters, several taxa of the gut microbiota were strongly correlated with inflammatory responses to SARS-CoV-2 infection. Positive correlations with lung histological scores and inflammatory cytokines were observed in Christensenellaceae, Desulfovibrioaceae, Flavobacteriaceae, and Peptococcaceae families, while negative correlations were seen in Butyricicoccaceae and Ruminococcaceae [9][2]. Similarly, in another study conducted on obese NASH hamsters, Blautia and Peptococcus were positively correlated with pro-inflammatory or pro-fibrotic profiles, whereas in lean hamsters Gordonibacter and Ileibacterium were negatively correlated with inflammatory profiles [10][3].
For human subjects, a study from Hong Kong showed that the COVID-19 patient cohort was found to have significant enrichment in Ruminococcus gnavus, Ruminococcus torques, and Bacteroides dorei, but lack Bifidobacterium adolescentis, Faecalibacterium prausnitzii, and Eubacterium rectale [11][4]. After adjusting for antibiotic use and patients’ age, F. prausnitzii and Bifidobacterium bifidum were found to have a significant negative correlation with COVID-19 severity. In addition, B. adolescentis, E. rectale, and F. prausnitzii, which were known to have immunomodulatory effects in the human gastrointestinal system, were negatively correlated with various immune markers. The depletion of these species may have contributed to overaggressive inflammation and even cytokine storms seen in severe COVID-19 cases. Another study from Japan showed similar findings [12][5]. In this study, the gut microbes enriched in the COVID-19 patient cohort, which included R. torques, were positively correlated with cytokines that were enriched during COVID-19, including those that were implicated with increased disease severity and cytokine storms. In contrast, gut microbes that were depleted in the COVID-19 cohort, which included B. adolescentis and E. rectale, were correlated with cytokines that were reduced during COVID-19, including CCL20 which was important for regulatory T cell migration. This suggested that the gut microbiota was involved in cytokine metabolism, which was in turn linked to inflammation and disease severity. In another Hong Kong study, 23 bacterial taxa in the baseline gut microbiome were found to be significantly associated with the severity of COVID-19, most of which were Firmicutes [13][6]. Again, F. prausnitzii were most negatively correlated with COVID-19 severity. A study conducted in Germany also found that the relative abundance of Faecalibacterium and Roseburia was lower in severe or critical COVID-19 cases [14][7]. Therefore, the gut microbiota and COVID-19 are very likely to have a dynamic relationship which can potentially form a vicious cycle with lasting effects.
The potential associations between the gut microbiota and COVID-19 severity were also indirectly implicated by studies that investigated the associations between the use of proton pump inhibitors (PPIs) and COVID-19 severity. These studies showed that PPI usage may increase the risk of COVID-19 positivity [15][8] and severity [16][9]. In an American study, there was a dose–response relationship in which those who took PPIs twice daily had a higher risk of being COVID-19-positive compared to just a single daily dose [15][8]. Lower-dose PPI use was also associated with lower odds of developing gastrointestinal symptoms of COVID-19 in individuals tested positive for COVID-19. In a Korean study, however, propensity score matching found that PPI use was not associated with COVID-19 positivity, but was associated with 79% increased risk of severe symptoms of COVID-19 [16][9]. PPIs alter the gut microbiome with significant increase in relative abundance of Enterococcus, Streptococcus, Staphylococcus, and the potentially pathogenic E. coli [17][10]. PPIs may also inhibit the activities of immune cells [18][11], increase the risk of enteric infections by suppressing gastric acid secretion [19][12], and alter immunomodulatory and anti-inflammatory effects [20][13]. In the context of the COVID-19 pandemic, the above effects of PPIs could have resulted in higher SARS-CoV-2 viral load in the GI tract, which contributed to more severe clinical outcomes [16][9].
3. COVID-19-Induced Gut Microbiome Alterations
COVID-19 could potentially induce alterations in the gut microbiota. Animal studies have demonstrated that SARS-CoV-2 infection in macaques was able to induce changes in gut microbiota and metabolome, which peaked at 10–13 days post infection [21][14]. In the hamster study mentioned previously, SARS-CoV-2 infection was characterized by the enrichment of deleterious bacterial taxa, including Enterobacteriaceae and Desulfovibrionaceae, as well as decreased relative abundance of several members of Ruminococcaceae and Lachnospiraceae families which included bacteria known to produce short-chain fatty acids (SCFAs) [9][2]. In obese hamsters, such SARS-CoV-2-induced changes in the gut microbiota could persist even longer [10][3]. This evidence suggested that the gut microbiota may be subjected to perturbations brought by body response to SARS-CoV-2.
A number of cross-sectional studies on human subjects have reported gut microbiota alterations in COVID-19 cases. In adult COVID-19 patients, there was decreased bacterial diversity in the fecal microbiome. Compared to healthy individuals, COVID-19 patients were observed to have reduced abundance of SCFA-producing bacteria, specifically Faecalibacterium, Eubacterium, Coprococcus, Ruminococcus, Lachnospira, and Roseburia, as well as increased abundance of opportunistic pathogens from Enterbacteriaceae families, specifically Enterococcus, Rothia and Lactobacillus. Notably, at the genus level, Bifidobacterium, Bacteroides, Streptococcus, and Enterococcus were most commonly reported to be enriched, while at the species level, Bifidobacterium longum and Ruminococcus torques were each reported in two separate studies. On the other hand, the genera Faecalibacterium, Eubacterium, Coprococcus, Bifidobacterium, and Clostridium were most commonly reported to be depleted, while at the species level Eubacterium hallii and Eubacterium rectale were each reported in two separate studies. In children with COVID-19, the genera Akkermansia and Bifidobacterium were most commonly found to be depleted. One study found increased abundance of Faecalibacterium, Fusobacterium, and Neisseria, as well as decreased abundance of Bifidobacterium, Blautia, Granulicatella, and Prevotella in infected children [22][15]. In another smaller study, the alteration of gut microbiome was predominated by Pseudomonas, and such alteration could sustain for up to almost two months. [23][16] Another study conducted in asymptomatic children reported decreased abundance of Bifidobacterium bifidum and Akkermansia muciniphila in SARS-CoV-2-positive stool samples [24][17]. These two species were linked to protection against inflammation in previous studies. In particular, multisystem inflammatory syndrome in children (MIS-C), which was associated with COVID-19, was commonly found to have enriched level of Clostridium. Studies have also found that these children had decreased Bifidobacterium [22][15] and Firmicutes including Faecalibacterium prausnitzii [25][18]. Of note, these studies were only observational in nature and were not able to show that the gut microbiota alterations were the result of SARS-CoV-2 infection as the baseline gut microbiome before SARS-CoV-2 infection was not profiled. Another interesting observation was that the gut microbiome composition in COVID-19 appeared to be distinct from that in patients with influenza [26,27][19][20] or viral pneumonia [13][6], which suggested that SARS-CoV-2 may have unique effects on the gut microbiome compared with other respiratory viruses [6][21].
Gut microbiota are also thought to be involved in the development of post-acute COVID-19 syndrome (PACS), or “long COVID”, which is associated with persistent respiratory, cardiovascular, neuropsychiatric, gastrointestinal, and dermatological symptoms [36,37][22][23]. Gut microbiome alterations observed in acute COVID-19 episodes could persist even after clearance of SARS-CoV-2 infection [11,13][4][6]. A study conducted in Hong Kong found that at 6 months, the gut microbiota of PACS patients were significantly depleted of Collinsella aerofaciens, F. prausnitzii, and Blautia obeum, as well as enriched with Ruminococcus gnavus and Bacteroides vulgatus [38][24]. In contrast, patients without PACS had fewer species altered, and the alterations were able to recover at 6 months. The relative abundance of Bifidobacterium pseudocatenulatum, F. prausnitzii, Roseburia inulinivorans, and Roseburia hominis, which were known to be beneficial to host immunity, had the largest inverse correlations with PACS at 6 months. PACS patients also had distinct gut metabolome compared to controls. Different patterns of gut microbiota were also seen in different PACS symptoms. Moreover, baseline Blautia wexlerae and Bifidobacterium longum had an inverse correlation with PACS at 6 months, whereas two Actinomyces species and Atopobium parvulum exhibited a positive correlation. Therefore, the dysbiosis of the gut microbiota may be involved in PACS development, though the exact extent of this involvement remains to be further investigated.
4. Potential Mechanisms Underlying the Gut Microbiota and SARS-CoV-2 Infection Outcomes
The gut microbiota were linked to SARS-CoV-2 infection in several ways (Figure 1). Firstly, animal studies have shown that the gut microbiota had a dynamic relationship with the angiotensin-converting enzyme [2][25] (ACE2), which serves as the cell entry receptor for SARS-CoV-2 [39][26] and is abundantly expressed in enterocytes along the small intestine [40][27]. On one hand, ACE2 could influence gut microbiota ecology through the deregulation of ACE2-regulated uptake of tryptophan in the small intestine, which in turn affects downstream manifestations [41][28]. On the other hand, gut microbiota could modulate ACE2 expression in the gut [42][29]. One study found that Coprobacillus was associated with the up-regulation of ACE2 while some Bacteroides bacterium and species such as Bifidobacterium longum were associated with the downregulation of ACE2 [43][30]. In particular, four of the Bacteroides species found in this study were shown to be negatively correlated with fecal SARS-CoV-2 load in human subjects [13][6]. This suggested that the gut microbiota can potentially influence the disease course and severity of COVID-19 by mediating ACE2-dependent SARS-CoV-2 entry. Further studies on human subjects, however, are needed to determine the exact mechanisms.
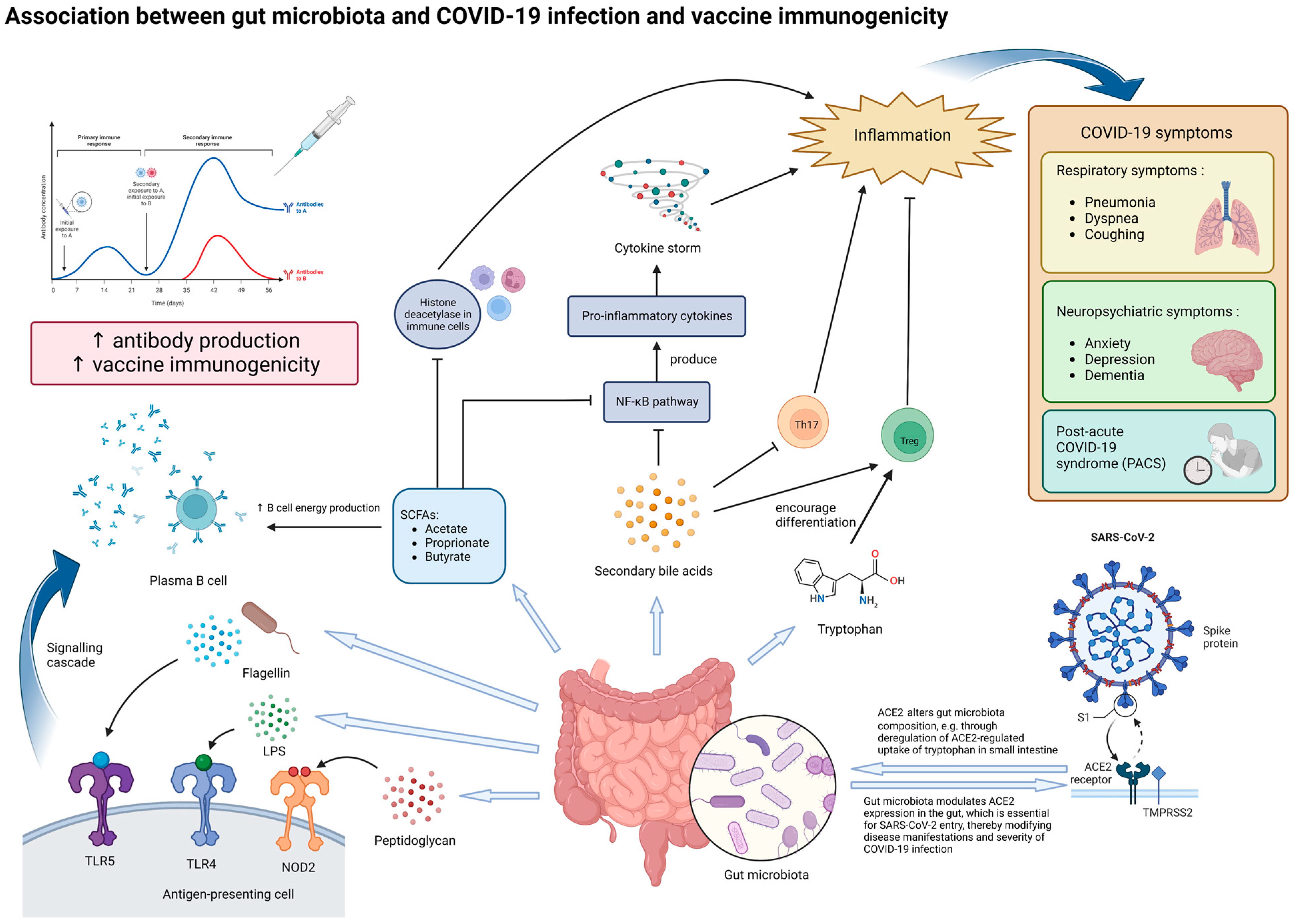
Figure 1. Potential mechanisms underlying the effect of gut microbiota on SARS-CoV-2 infection and vaccine immunogenicity. The gut microbiota and its metabolites, particularly those with immunomodulatory properties, can influence both the manifestations of COVID-19 and vaccine immunogenicity. In the context of COVID-19, dysbiosis of the gut microbiota may increase the severity of inflammation and various symptoms through modulating ACE2 expression in enterocytes and altered secretion of immunomodulatory molecules, such as tryptophan, SCFAs and secondary bile acids. Dysbiosis may potentially contribute to the production of cytokine storms, which produce more severe symptoms. In the long run, dysbiosis may be associated with persistent COVID-19 symptoms and inflammation, termed as post-acute COVID-19 syndrome (PACS). In the context of vaccine immunogenicity, lipopolysaccharides (LPSs), flagellin, peptidoglycan, and SCFAs secreted by the gut microbiota can enhance antibody production to vaccination by plasma B cells, thereby improving vaccine immunogenicity. Abbreviations: ACE2, angiotensin-converting enzyme 2; LPS, lipopolysaccharide; NOD2, nucleotide-binding oligomerization domain-containing protein 2; TLR-4, Toll-like receptor 4; TLR-5, Toll-like receptor 5; SCFA, short-chain fatty acid; NF-κB, nuclear factor-κB; Th17, T helper 17 cells; Treg, regulatory T cells.
In addition, metabolites produced by the gut microbiota are also likely to play important roles in the immune response against SARS-CoV-2 infection. Examples of such metabolites include tryptophan, SCFAs, and secondary bile acids. Tryptophan and the metabolites derived from it (such as kynurenine and indoles) are important in changing the functions of various immune cells (such as regulatory T cells) and mediating inflammation [44][31]. A decrease in tryptophan was found to be associated with more severe COVID-19 symptoms [45,46][32][33]. This suggests that tryptophan metabolism may be another one of the links between the gut microbiota and COVID-19 development.
SCFAs produced by the gut microbiota, such as acetate, propionate, and butyrate, can enter systemic circulation through passive diffusion or active transport by the gut epithelial cells, where they then modulate local and systemic inflammation and immune responses [47][34]. In particular, butyrate has been observed to support the integrity of the gut barrier and gut homeostasis and can exert anti-inflammatory effects through the inhibition of histone deacetylase in various immune cells and the activation of nuclear factor-κB (NF-κB), which in turn reduces the production of proinflammatory cytokines [48][35]. As mentioned previously, butyrate-producing species, especially F. prausnitzii, have been frequently observed to be depleted in COVID-19 patients, especially in those with more severe and persistent symptoms [6,11,13,14,25,38][4][6][7][18][21][24]. Indeed, F. prausnitzii has also been observed to be reduced in other inflammatory diseases, such as Crohn’s disease [49][36]. These observations support the notion that alterations in SCFA production, particularly butyrate, due to gut microbiota dysbiosis may exacerbate inflammation and thus the severity of COVID-19. Supplementation of the gut microbe species involved and SCFAs themselves can be a potential therapeutic strategy as an adjuvant to reduce the symptoms brought by COVID-19. In fact, this strategy has been tested on animal models for the treatment of inflammatory bowel diseases, such as colitis in rat models, with promising results [50,51][37][38].
Similarly, secondary bile acids have also been shown to be able to inhibit NF-κB signaling pathways, inhibit IL-17 expressing helper T cells, and enhance differentiation of regulatory T cells [52][39]. In the context of COVID-19, secondary bile acids were found to be significantly associated with the progression of respiratory failure and patient’s survival [53][40]. Ursodeoxycholic acid has also been suggested as a therapeutic agent for the prevention of cytokine storms in COVID-19 management by inhibiting the production of pro-inflammatory cytokines [54,55][41][42]. In line with this, one study found that the abundance of Collinsella, a major producer of ursodeoxycholate, was inversely correlated with COVID-19 mortality [56][43]. Collinsella had also been found to be significantly depleted in patients with PACS [38][24]. Therefore, secondary bile acids produced by the gut microbiota may very likely be involved in modulating the presentation of COVID-19 in different individuals. These metabolites were also important in mediating the crosstalk between the gut and other organs. One such crosstalk that is important in COVID-19 is that between the gut and the lungs, also known as the “gut-lung axis”. Through this axis, intact or fragmented gut bacteria, as well as their metabolites such as SCFAs, can cross the intestinal barrier and modulate the local immune response in the lungs via systemic circulation [57][44]. Studies have also shown that the gut microbiota could influence the expression of type I interferon receptors in respiratory epithelial cells, thereby mediating the secretion of IFNɑ and IFNβ and restricting viral replication [58][45]. Through the gut–lung axis, gut microbiota dysbiosis may potentially influence respiratory symptoms in COVID-19. In addition, the crosstalk between the gut and the brain, termed the “gut-brain axis”, may have been involved in the development of neuropsychiatric symptoms in COVID-19. Dysbiosis of the gut microbiota has been observed in various neuropsychiatric disorders that may be present in COVID-19 patients, such as anxiety, depression, and dementia [47][34]. SCFAs produced by the gut microbiota can bind to G protein-coupled receptors in the brain to modulate neuronal activity and mediate brain immunity [47,59][34][46]. Therefore, SCFA deficiency due to dysbiosis could have contributed to inflammation in the brain and other neuropsychiatric complications seen in COVID-19.
References
- Sarkar, A.; Harty, S.; Moeller, A.H.; Klein, S.L.; Erdman, S.E.; Friston, K.J.; Carmody, R.N. The gut microbiome as a biomarker of differential susceptibility to SARS-CoV-2. Trends Mol. Med. 2021, 27, 1115–1134.
- Sencio, V.; Machelart, A.; Robil, C.; Benech, N.; Hoffmann, E.; Galbert, C.; Deryuter, L.; Heumel, S.; Hantute-Ghesquier, A.; Flourens, A.; et al. Alteration of the gut microbiota following SARS-CoV-2 infection correlates with disease severity in hamsters. Gut Microbes 2022, 14, 2018900.
- Sencio, V.; Benech, N.; Robil, C.; Deruyter, L.; Heumel, S.; Machelart, A.; Sulpice, T.; Lamaziere, A.; Grangette, C.; Briand, F.; et al. Alteration of the gut microbiota’s composition and metabolic output correlates with COVID-19-like severity in obese NASH hamsters. Gut Microbes 2022, 14, 2100200.
- Yeoh, Y.K.; Zuo, T.; Lui, G.C.; Zhang, F.; Liu, Q.; Li, A.Y.; Chung, A.C.; Cheung, C.P.; Tso, E.Y.; Fung, K.S.; et al. Gut microbiota composition reflects disease severity and dysfunctional immune responses in patients with COVID-19. Gut 2021, 70, 698–706.
- Nagata, N.; Takeuchi, T.; Masuoka, H.; Aoki, R.; Ishikane, M.; Iwamoto, N.; Sugiyama, M.; Suda, W.; Nakanishi, Y.; Terada-Hirashima, J.; et al. Human Gut Microbiota and Its Metabolites Impact Immune Responses in COVID-19 and Its Complications. Gastroenterology 2022, 164, 272–288.
- Zuo, T.; Zhang, F.; Lui, G.C.Y.; Yeoh, Y.K.; Li, A.Y.L.; Zhan, H.; Wan, Y.; Chung, A.C.K.; Cheung, C.P.; Chen, N.; et al. Alterations in Gut Microbiota of Patients With COVID-19 During Time of Hospitalization. Gastroenterology 2020, 159, 944–955e8.
- Reinold, J.; Farahpour, F.; Fehring, C.; Dolff, S.; Konik, M.; Korth, J.; van Baal, L.; Hoffmann, D.; Buer, J.; Witzke, O.; et al. A Pro-Inflammatory Gut Microbiome Characterizes SARS-CoV-2 Infected Patients and a Reduction in the Connectivity of an Anti-Inflammatory Bacterial Network Associates with Severe COVID-19. Front. Cell Infect. Microbiol. 2021, 11, 747816.
- Almario, C.V.; Chey, W.D.; Spiegel, B.M.R. Increased Risk of COVID-19 Among Users of Proton Pump Inhibitors. Am. J. Gastroenterol. 2020, 115, 1707–1715.
- Lee, S.W.; Ha, E.K.; Yeniova, A.O.; Moon, S.Y.; Kim, S.Y.; Koh, H.Y.; Yang, J.M.; Jeong, S.J.; Moon, S.J.; Cho, J.Y.; et al. Severe clinical outcomes of COVID-19 associated with proton pump inhibitors: A nationwide cohort study with propensity score matching. Gut 2021, 70, 76–84.
- Imhann, F.; Bonder, M.J.; Vich Vila, A.; Fu, J.; Mujagic, Z.; Vork, L.; Tigchelaar, E.F.; Jankipersadsing, S.A.; Cenit, M.C.; Harmsen, H.J.M.; et al. Proton pump inhibitors affect the gut microbiome. Gut 2016, 65, 740–748.
- Laheij, R.J.; Sturkenboom, M.C.; Hassing, R.J.; Dieleman, J.; Stricker, B.H.; Jansen, J.B. Risk of community-acquired pneumonia and use of gastric acid-suppressive drugs. JAMA 2004, 292, 1955–1960.
- Vaezi, M.F.; Yang, Y.X.; Howden, C.W. Complications of Proton Pump Inhibitor Therapy. Gastroenterology 2017, 153, 35–48.
- Trifan, A.; Stanciu, C.; Girleanu, I.; Stoica, O.C.; Singeap, A.M.; Maxim, R.; Chiriac, S.A.; Ciobica, A.; Boiculese, L. Proton pump inhibitors therapy and risk of Clostridium difficile infection: Systematic review and meta-analysis. World J. Gastroenterol. 2017, 23, 6500–6515.
- Sokol, H.; Contreras, V.; Maisonnasse, P.; Desmons, A.; Delache, B.; Sencio, V.; Machelart, A.; Brisebarre, A.; Humbert, L.; Deryuter, L.; et al. SARS-CoV-2 infection in nonhuman primates alters the composition and functional activity of the gut microbiota. Gut Microbes 2021, 13, 1893113.
- Romani, L.; Del Chierico, F.; Macari, G.; Pane, S.; Ristori, M.V.; Guarrasi, V.; Gardini, S.; Pascucci, G.R.; Cotugno, N.; Perno, C.F.; et al. The Relationship Between Pediatric Gut Microbiota and SARS-CoV-2 Infection. Front. Cell Infect. Microbiol. 2022, 12, 908492.
- Xu, R.; Liu, P.; Zhang, T.; Wu, Q.; Zeng, M.; Ma, Y.; Jin, X.; Xu, J.; Zhang, Z.; Zhang, C. Progressive deterioration of the upper respiratory tract and the gut microbiomes in children during the early infection stages of COVID-19. J. Genet. Genomics 2021, 48, 803–814.
- Nashed, L.; Mani, J.; Hazrati, S.; Stern, D.B.; Subramanian, P.; Mattei, L.; Bittinger, K.; Hu, W.; Levy, S.; Maxwell, G.L.; et al. Gut microbiota changes are detected in asymptomatic very young children with SARS-CoV-2 infection. Gut 2022, 71, 2371–2373.
- Suskun, C.; Kilic, O.; Yilmaz Ciftdogan, D.; Guven, S.; Karbuz, A.; Ozkaya Parlakay, A.; Kara, Y.; Kacmaz, E.; Sahin, A.; Boga, A.; et al. Intestinal microbiota composition of children with infection with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and multisystem inflammatory syndrome (MIS-C). Eur. J. Pediatr. 2022, 181, 3175–3191.
- Gu, S.; Chen, Y.; Wu, Z.; Chen, Y.; Gao, H.; Lv, L.; Guo, F.; Zhang, X.; Luo, R.; Huang, C.; et al. Alterations of the Gut Microbiota in Patients with Coronavirus Disease 2019 or H1N1 Influenza. Clin. Infect. Dis. 2020, 71, 2669–2678.
- Tao, W.; Zhang, G.; Wang, X.; Guo, M.; Zeng, W.; Xu, Z.; Cao, D.; Pan, A.; Wang, Y.; Zhang, K.; et al. Analysis of the intestinal microbiota in COVID-19 patients and its correlation with the inflammatory factor IL-18. Med. Microecol. 2020, 5, 100023.
- Zhang, F.; Lau, R.I.; Liu, Q.; Su, Q.; Chan, F.K.L.; Ng, S.C. Gut microbiota in COVID-19: Key microbial changes, potential mechanisms and clinical applications. Nat. Rev. Gastroenterol. Hepatol. 2022, 1–15.
- Nalbandian, A.; Sehgal, K.; Gupta, A.; Madhavan, M.V.; McGroder, C.; Stevens, J.S.; Cook, J.R.; Nordvig, A.S.; Shalev, D.; Sehrawat, T.S.; et al. Post-acute COVID-19 syndrome. Nat. Med. 2021, 27, 601–615.
- Montani, D.; Savale, L.; Noel, N.; Meyrignac, O.; Colle, R.; Gasnier, M.; Corruble, E.; Beurnier, A.; Jutant, E.M.; Pham, T.; et al. Post-acute COVID-19 syndrome. Eur. Respir. Rev. 2022, 31, 210185.
- Liu, Q.; Mak, J.W.Y.; Su, Q.; Yeoh, Y.K.; Lui, G.C.-Y.; Ng, S.S.S.; Zhang, F.; Li, A.Y.L.; Lu, W.; Hui, D.S.-C.; et al. Gut microbiota dynamics in a prospective cohort of patients with post-acute COVID-19 syndrome. Gut 2022, 71, 544–552.
- Mao, R.; Qiu, Y.; He, J.S.; Tan, J.-Y.; Li, X.-H.; Liang, J.; Shen, J.; Zhu, L.-R.; Chen, Y.; Iacucci, M.; et al. Manifestations and prognosis of gastrointestinal and liver involvement in patients with COVID-19: A systematic review and meta-analysis. Lancet Gastroenterol. Hepatol. 2020, 5, 667–678.
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273.
- Hamming, I.; Timens, W.; Bulthuis, M.L.; Lely, A.T.; Navis, G.J.; van Goor, H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J. Pathol. 2004, 203, 631–637.
- Hashimoto, T.; Perlot, T.; Rehman, A.; Trichereau, J.; Ishiguro, H.; Paolino, M.; Sigl, V.; Hanada, T.; Hanada, R.; Lipinski, S.; et al. ACE2 links amino acid malnutrition to microbial ecology and intestinal inflammation. Nature 2012, 487, 477–481.
- Yang, T.; Chakraborty, S.; Saha, P.; Mell, B.; Cheng, X.; Yeo, J.Y.; Mei, X.; Zhou, G.; Mandal, J.; Golonka, R.; et al. Gnotobiotic Rats Reveal That Gut Microbiota Regulates Colonic mRNA of Ace2, the Receptor for SARS-CoV-2 Infectivity. Hypertension 2020, 76, e1–e3.
- Geva-Zatorsky, N.; Sefik, E.; Kua, L.; Pasman, L.; Tan, T.G.; Ortiz-Lopez, A.; Yanortsang, T.B.; Yang, L.; Jupp, R.; Mathis, D.; et al. Mining the Human Gut Microbiota for Immunomodulatory Organisms. Cell 2017, 168, 928–943e11.
- Sorgdrager, F.J.H.; Naude, P.J.W.; Kema, I.P.; Nollen, E.A.; De Deyn, P.P. Tryptophan Metabolism in Inflammaging: From Biomarker to Therapeutic Target. Front. Immunol. 2019, 10, 2565.
- Almulla, A.F.; Supasitthumrong, T.; Tunvirachaisakul, C.; Algon, A.A.A.; Al-Hakeim, H.K.; Maes, M. The tryptophan catabolite or kynurenine pathway in COVID-19 and critical COVID-19: A systematic review and meta-analysis. BMC Infect. Dis. 2022, 22, 615.
- Blackett, J.W.; Sun, Y.; Purpura, L.; Margolis, K.G.; Elkind, M.S.; O’Byrne, S.; Wainberg, M.; Abrams, J.A.; Wang, H.H.; Chang, L.; et al. Decreased Gut Microbiome Tryptophan Metabolism and Serotonergic Signaling in Patients with Persistent Mental Health and Gastrointestinal Symptoms After COVID-19. Clin. Transl. Gastroenterol. 2022, 13, e00524.
- Wlodarczyk, J.; Czerwinski, B.; Fichna, J. Short-chain fatty acids-microbiota crosstalk in the coronavirus disease (COVID-19). Pharmacol. Rep. 2022, 74, 1198–1207.
- Siddiqui, M.T.; Cresci, G.A.M. The Immunomodulatory Functions of Butyrate. J. Inflamm. Res. 2021, 14, 6025–6041.
- Sokol, H.; Pigneur, B.; Watterlot, L.; Lakhdari, O.; Bermúdez-Humaran, L.G.; Gratadoux, J.-J.; Blugeon, S.; Bridonneau, C.; Furet, J.-P.; Corthier, G.; et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc. Natl. Acad. Sci. USA 2008, 105, 16731–16736.
- Qiu, X.; Zhang, M.; Yang, X.; Hong, N.; Yu, C. Faecalibacterium prausnitzii upregulates regulatory T cells and anti-inflammatory cytokines in treating TNBS-induced colitis. J. Crohns Colitis 2013, 7, e558–e568.
- Zhang, M.; Qiu, X.; Zhang, H.; Yang, X.; Hong, N.; Yang, Y.; Chen, H.; Yu, C. Faecalibacterium prausnitzii inhibits interleukin-17 to ameliorate colorectal colitis in rats. PLoS ONE 2014, 9, e109146.
- Gautier, T.; Gall, S.D.-L.; Sweidan, A.; Tamanai-Shacoori, Z.; Jolivet-Gougeon, A.; Loréal, O.; Bousarghin, L. Next-Generation Probiotics and Their Metabolites in COVID-19. Microorganisms 2021, 9, 941.
- Stutz, M.R.; Dylla, N.P.; Pearson, S.D.; Lecompte-Osorio, P.; Nayak, R.; Khalid, M.; Adler, E.; Boissiere, J.; Lin, H.; Leiter, W.; et al. Immunomodulatory fecal metabolites are associated with mortality in COVID-19 patients with respiratory failure. Nat. Commun. 2022, 13, 6615.
- Abdulrab, S.; Al-Maweri, S.; Halboub, E. Ursodeoxycholic acid as a candidate therapeutic to alleviate and/or prevent COVID-19-associated cytokine storm. Med. Hypotheses 2020, 143, 109897.
- Subramanian, S.; Iles, T.; Ikramuddin, S.; Steer, C.J. Merit of an Ursodeoxycholic Acid Clinical Trial in COVID-19 Patients. Vaccines 2020, 8, 320.
- Hirayama, M.; Nishiwaki, H.; Hamaguchi, T.; Ito, M.; Ueyama, J.; Maeda, T.; Kashihara, K.; Tsuboi, Y.; Ohno, K. Intestinal Collinsella may mitigate infection and exacerbation of COVID-19 by producing ursodeoxycholate. PLoS ONE 2021, 16, e0260451.
- Enaud, R.; Prevel, R.; Ciarlo, E.; Beaufils, F.; Wieërs, G.; Guery, B.; Delhaes, L. The Gut-Lung Axis in Health and Respiratory Diseases: A Place for Inter-Organ and Inter-Kingdom Crosstalks. Front. Cell. Infect. Microbiol. 2020, 10, 9.
- de Oliveira, G.L.V.; Oliveira, C.N.S.; Pinzan, C.F.; de Salis, L.V.V.; Cardoso, C.R.D.B. Microbiota Modulation of the Gut-Lung Axis in COVID-19. Front. Immunol. 2021, 12, 635471.
- Sajdel-Sulkowska, E.M. Neuropsychiatric Ramifications of COVID-19: Short-Chain Fatty Acid Deficiency and Disturbance of Microbiota-Gut-Brain Axis Signaling. Biomed. Res. Int. 2021, 2021, 7880448.
More