T 辅助细胞helper 22 (Th22) 细胞是一种新定义的cells, a newly defined CD4+ T 细胞谱系,其特征在于其独特的细胞因子谱,主要由-cell lineage, are characterized by their distinct cytokine profile, which primarily consists of IL-13、, IL-22 和and TNF-α 组成。Th22 细胞表达广泛的趋化因子受体,例如 CCR4、CCR6 和. Th22 cells express a wide spectrum of chemokine receptors, such as CCR4, CCR6 and CCR10。Th22 细胞分泌的主要效应分子是. The main effector molecule secreted by Th22 cells is IL-22,它是, a member of the IL-10 家族的一员,它通过与 family, which acts by binding to IL-22R 结合并触发复杂的下游信号系统发挥作用。已发现and triggering a complex downstream signaling system. Th22 细胞和cells and IL-22 在人体免疫中发挥着不同的作用。在预防 HIV 和流感等感染的进展方面,have been found to play variable roles in human immunity. In preventing the progression of infections such as HIV and influenza, Th22/IL-22 表现出保护性抗炎特性,其有害的促炎活性已被证明会加剧其他疾病,包括乙型肝炎和幽门螺杆菌感染。exhibited protective anti-inflammatory characteristics, and their deleterious proinflammatory activities have been demonstrated to exacerbate other illnesses, including hepatitis B and Helicobacter pylori infection.
- Th22 cells
- IL-22
- immunity
- infection
一、简介1. Introduction
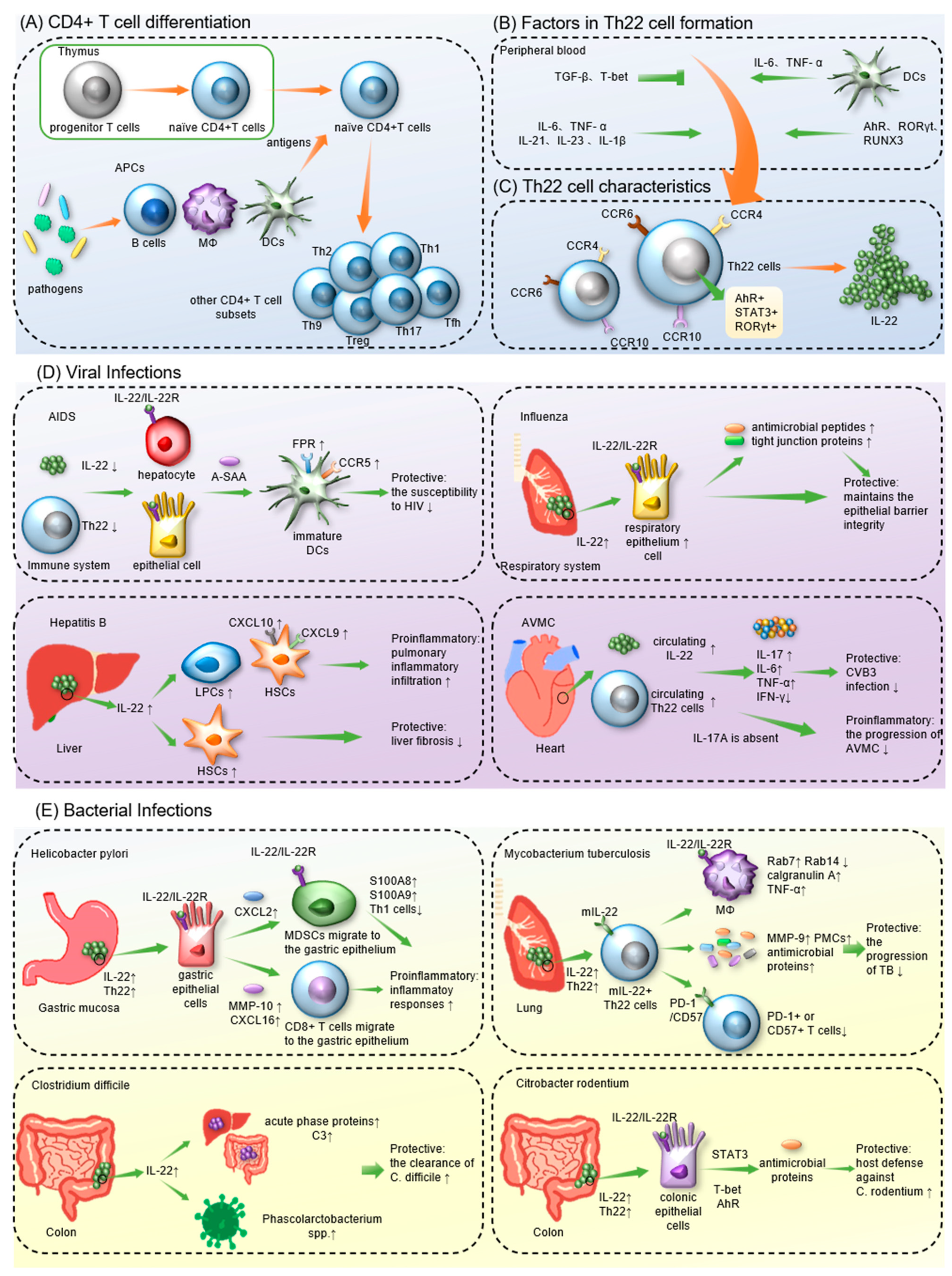
1.1. The Discovery of IL-22 and Th22 Cells
1.2. Factors in the Formation of Th22 cells and IL-22
1.3. The Effects of Th22/IL-22
2. Th22 Cells in Infectious Diseases
2.1. Th22 Cells in Viral Infections
2.1.1. COVID-19
COVID-19 is pneumonia induced by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Studies in the context of infectious diseases have focused on the pathogenic mechanisms of COVID-19 since its outbreak in 2019. Hoffmann et al. proposed that COVID-19 shares some similar symptoms with influenza and respiratory syncytial virus (RSV)-induced pneumonia [56]. According to previous studies, IL-22/Th22 is protective against influenza and RSV pneumonia [54,57,58][54][57][58] and may exert a similar effect against COVID-19. Among patients with fulminant COVID-19-related myocarditis, some met the criteria for multisystem inflammatory syndrome (MIS-A+), whereas the rest did not (MIS-A-) [59]. Compared to the MIS-A- group, MIS-A+ patients showed higher expression of IL-22, a better prognosis and lower mortality [59]. These results suggest that IL-22 may have a protective and antiviral effect in MIS-A+ COVID-19 patients. A novel study noted that abnormal dynamic IL-22R1 expression on blood myeloid cells and CD4+ T cells is a characteristic of SARS-CoV-2 infection [60]. IL-22R1 expression on myeloid cells is discriminative for the severity of COVID-19 [60]. However, COVID-19 patients with different prognoses have similar IL-22 levels, suggesting that IL-22 does not affect the outcomes of SARS-CoV-2 infection [61]. Furthermore, the number of IL-22R1-expressing myeloid cells is correlated with the plasma levels of COVID-19-related immune mediators [60]. During the acute phase of COVID-19, the immune response leads to a dramatic increase in several cytokines, including IL-22, which is mainly produced by Th22 cells [62,63][62][63]. This process is called cytokine release syndrome (CRS) and contributes to fatal complications, such as multiple organ failure and acute respiratory distress syndrome [64,65][64][65]. This result indicated that IL-22R1+ myeloid cells may participate in the cascade, leading to CRS and promoting the deterioration of COVID-19. This study also suggested that the IL-22-induced signaling pathway switches from protective to pathogenic as the disease progresses [60]. Therefore, IL-22/Th22 cells may play a critical role in the pathological process of COVID-19, but the detailed mechanism still awaits further research.2.1.2. AIDS
Acquired immunodeficiency syndrome (AIDS), an infectious illness that arises from infection with human immunodeficiency virus (HIV), is known for its high mortality and prolonged course [66]. In comparison with healthy controls and HIV-infected patients, more acute-phase serum amyloid A (A-SAA) and IL-22 are produced in HIV-exposed but uninfected individuals (EUs) [67,68][67][68]. IL-22 has been confirmed to promote the expression of A-SAA in epithelial cells and liver cells [47,49,69][47][49][69]. Moreover, Misse’ et al. coincubated A-SAA with immature DCs in vitro for further exploration. A-SAA is an agonist of the formyl peptide receptor (FPR) and enhances FPR expression on DCs [67]. The FPR promoted the phosphorylation of CCR5 and decreased the expression of CCR5 on DCs, resulting in decreased susceptibility of DCs to HIV [67]. Therefore, it was suggested that the high resistance of EUs to HIV may be associated with IL-22-induced A-SAA. However, the higher sensitivity of Th22 cells to HIV compared to other CD4+ T cell subsets is attributed to the high expression of CCR5 and α4β7 on Th22 cells [70]. Both CCR5 and α4β7 are the main binding sites for HIV. Therefore, the counts and functional scores of Th22 cells are decreased significantly in the sigmoid colon of HIV patients [71,72][71][72]. Moreover, since Th22/IL-22 limits the translocation of commensal bacteria into the systemic circulation [73], Th22 depletion in HIV patients not only causes impairment of the intestinal barrier but also moves intestinal bacteria to the lamina propria [71,72][71][72]. The translocated intestinal bacteria eventually enter the circulation and cause systemic immune activation, which is key to the pathogenesis of HIV [72]. Furthermore, viral load in HIV-infected individuals is negatively associated with the level of serum IL-22, and one study indicated that IL-22 may inhibit HIV replication by regulating C-reactive protein (CRP) and IL-10 [74]. Compared to HIV patients with inflammatory disease coinfection, patients without inflammatory diseases showed lower neutrophil activation and a surplus of IL-22 expression [75]. This result suggests that IL-22 controls HIV-related inflammatory injury by regulating neutrophil hyperactivation [75]. In patients infected with HIV-2, a spontaneously attenuating HIV strain, the expression of CCL20 and CCL28 was significantly increased in the sigmoid colon, leading to local recruitment of Th22 cells [76]. Therefore, Th22 cells counteract intestinal CD4+ T-cell exhaustion and maintain intestinal mucosal integrity. This finding may explain why HIV-2 infection is less dangerous than HIV-1 infection. In addition, mucosa-associated constant T-cell (MAIT) levels were positively correlated with Th22 cell frequency in HIV-infected children [77]. Since innate immune responses are effectively regulated by MAITs [78[78][79],79], Th22 cells may also be antiviral in HIV-infected children by mediating the level of MAITs. Moreover, compared to HIV-infected individuals with immune responses (IRs), patients without immune responses (INRs) expressed more IL-22 in the colon and showed more severe mucosal damage [80]. After receiving antiretroviral therapy (ART), INRs had poorer immune recovery than IRs [80], indicating that the dysregulation of IL-22 frequency may be responsible for the poor prognosis of INRs. Hence, although Th22 cells are downregulated in HIV patients, they modulate mucosal immunity and acute phase protein expression to suppress the progression of AIDS.2.1.3. Hepatitis
Viral hepatitis is typically caused by infection with hepatitis B or C viruses [81,82][81][82]. Both hepatitis B and C can increase the risk of cirrhosis and hepatocellular carcinoma [81,83][81][83]. According to previous studies, IL-22 showed weak anti-HBV effects and no direct antiviral activity against hepatitis C virus (HCV) [84]. Surprisingly, IL-22-/- mice showed increased susceptibility to inflammatory liver damage, demonstrating that IL-22 protects hepatocytes through mechanisms other than direct virus killing [85]. In HBV-infected patients, both intrahepatic and serum IL-22 expression levels are significantly elevated [86]. It has been confirmed that through the STAT3 pathway, IL-22 stimulates the proliferation of liver stem/progenitor cells (LPCs), cells participating in liver inflammatory responses [86]. Moreover, IL-22 production in hepatitis B patients is positively related to the HBV load [87]. By increasing CXCL9 and CXCL10 expression on hepatic stellate cells (HSCs), IL-22 also promotes inflammatory cell accumulation in the liver, leading to increased liver damage and hepatic fibrosis [88]. As a result, IL-22 may be both proinflammatory and profibrogenic in hepatitis B. In addition, elevated circulating levels of Th22 correlated with the severity of HBV-associated acute–chronic liver failure (HBV-ACLF), suggesting that Th22 cells are a negative predictor of prognosis in HBV-ACLF [89]. Nevertheless, according to Kong et al., IL-22 can activate STAT3 and p53 to induce senescence in HSCs [90]. IL-22 levels also negatively correlate with the development of liver fibrosis [91]. Therefore, the proinflammatory or protective effect of IL-22 during HBV infection may vary according to different disease states [92,93][92][93]. For hepatitis C, IL-22 mRNA and Th22 cell levels were elevated in the livers of patients with chronic hepatitis C (CHC) compared to controls [84[84][93][94],93,94], indicating that Th22 cells may be recruited to the liver by intrahepatic chemokines [94]. Furthermore, IL-22 induces the proliferation of HSCs, and the Th22 cell level is positively related to the progression of CHC to cirrhosis, suggesting that Th22/IL-22 facilitates HCV-related liver fibrosis [95,96][95][96]. However, an in vitro study confirmed that two variants of the gene encoding IL-22BP are correlated with HCV-mediated liver fibrosis and cirrhosis. It has also been proven that high production of IL-22 is correlated with protective immune responses to hepatitis C [51]. Furthermore, during the progression of hepatitis C to cirrhosis, the ratio of IL-22BP/IL-22 increases with the stage of liver fibrosis and peaks at the time of cirrhosis [97]. Hence, the administration of IL-22BP inhibitors, such as IL-18, prostaglandin E2 (PGE2), NLRP3 and NLRP6 inflammasomes [98,99,100][98][99][100], may be a promising therapy for liver fibrosis [51]. This conclusion was questioned by Wu et al. [101]. They stated that the correlation between IL-22 and protective responses in vitro may not be available in vivo because physiological IL-22 levels in the liver cannot be accurately measured in vitro [101], which means that the exact role of IL-22 in hepatitis C remains controversial.2.1.4. Influenza
Influenza is a highly infectious disease that primarily invades the respiratory system. By attacking lung epithelial cells, influenza viruses can destroy the pulmonary epithelial barrier and lead to abnormal gas exchange and pulmonary effusion [102]. After infection with influenza A virus (IAV), IL-22 levels in the lung tissues of patients are significantly increased and can gradually return to normal as the disease improves [103,104,105][103][104][105]. Moreover, the lung injury caused by H1N1 IAV is more severe in IL-22-/- mice than in controls [57]. These findings suggest that IL-22 may have a protective role during influenza infection. A study showed that some IAV-infected mice can inhibit IL-22 production by producing type I interferons (I-IFN), and they were at a higher risk of secondary infection with S. aureus [106]. In mice infected with IAV, IL-22 protects the integrity of the epithelial barrier and inhibits secondary infection by inducing antimicrobial peptides and intercellular junction proteins expressed in the respiratory epithelium [107]. Tight junction proteins promote fluid efflux to alleviate pulmonary effusion in influenza A patients [108,109][108][109]. In addition, IL-22 has also been proven to inhibit IAV-induced lung epithelial cell necrosis, suppress inflammatory responses and promote bronchial epithelial cell regeneration [104,105,110][104][105][110]. Since IL-22 can be blocked by IL-22BP, IL-22BP inhibitors have been assumed to be effective at improving influenza prognosis [54]. Nevertheless, in contrast to the upregulated IL-22 expression in patients with a mild infection, the IL-22 levels were decreased remarkably in patients with severe influenza A [111]. Accordingly, IL-22 protects the respiratory system in IAV-infected patients and can be dysregulated with disease progression.2.1.5. Acute Viral Myocarditis
Acute viral myocarditis (AVMC) is nonspecific interstitial myocardial inflammation. Coxsackievirus B3 (CVB3) infection is the major cause [112]. Compared to the controls, circulating Th22 cells and IL-22 were significantly upregulated in mice infected with CVB3 [113]. Furthermore, in CVB3-infected mice, the anti-IL-22 antibody reduced the antiviral IFN-γ production while increasing the levels of proinflammatory cytokines such as IL-17, IL-6 and TNF-α [113]. Consequently, the antibody resulted in the deterioration of AVMC, demonstrating that Th22/IL-22 can regulate the expression of cytokines and improve antiviral activity and prognosis during CVB3 infection. If left untreated, AVMC can progress to dilated cardiomyopathy (DCM) [112]. Using animal models of CVB3-induced chronic myocarditis and DCM, studies have revealed that Th22/IL-22 also has a protective role in chronic viral myocarditis and that IL-22 can inhibit cardiac fibrosis [114]. Therefore, Th22/IL-22 may be a promising target for treating coxsackievirus-induced acute viral myocarditis, chronic viral myocarditis, and DCM. Nevertheless, in IL-17A-/- mice with AVMC, IL-22 neutralization contributes to improving acute myocarditis while increasing viral replication at the same time [115]. This result suggests that when IL-17A is absent, IL-22 can exacerbate the progression of AVMC and inhibit CVB3 replication.2.1.6. Other Viral Infections
Hand, foot, and mouth disease (HFMD) occurs mainly in children infected with coxsackievirus A16 (CV-A16) or enterovirus 71 (EV-71), leading to characteristic herpes on the hand, foot, mouth, and buttock [116]. In the acute phase of EV-71-induced HFMD, levels of both circulating Th22 cells and IL-22 are higher than in the convective phase. In addition, HFMD patients complicated with viral encephalitis have higher levels of IL-22 than patients with HFMD alone [117]. This result suggests that Th22/IL-22 may be crucial in the progression of HFMD [118]. Strong bronchitis in infants [119,120][119][120] and pneumonia in elderly or low-resistance patients [121] are often caused by respiratory syncytial virus (RSV) infection. The robust Th22 response during the acute phase of RSV infection predicts prolonged hospitalization, indicating a negative effect of Th22/IL-22 on RSV infection [122]. Nevertheless, contrary to the pathogenic effect of endogenous IL-22, exogenous IL-22 activates the STAT3 pathway in RSV-infected cells to promote apoptosis and inhibit viral replication [58]. Therefore, the administration of IL-22 may be effective in RSV therapy. Warts are a contagious skin disease primarily caused by human papillomavirus (HPV) [123]. In comparison with healthy controls, patients with warts produced more IL-22 in the serum, and their IL-22 levels were positively related to the wart counts, suggesting that IL-22 is involved in the immune response against HPV [124]. Yellow fever (YF) is endemic in the tropics and is characterized by acute fever, jaundice, and proteinuria [125,126][125][126]. It is typically induced by infection with yellow fever virus (YFV) [125]. Compared with healthy controls, IL-22 production by liver parenchymal cells from YF patients is significantly upregulated [127]. In addition, Mendes et al. speculated that IL-22 participates in M2 macrophage-mediated organ repair and the immune escape mechanism of YFV [127]. Consequently, Th22/IL-22 has both a protective and a proinflammatory role in viral infections, which is greatly affected by factors such as the subtype of virus, the severity of infection and the presence of IL-17A.2.2. Th22 Cells in Bacterial Infections
2.2.1. Mycobacterium tuberculosis
Mycobacterium tuberculosis (MTB) is a common pathogen that is primarily transmitted via the respiratory tract [128]. Compared to healthy controls, TB patients had lower levels of IL-22 and IL-22+ T cells in their plasma [129,130][129][130]. Th22 cells are the main IL-22 producers during MTB infection, but other subsets, such as Th1 cells, CD8+ T cells, and NKT cells, also secrete IL-22 [131,132,133][131][132][133]. In addition, the bronchoalveolar lavage fluid (BALF) of pulmonary TB patients contains a large amount of IL-22 at significantly higher levels than the corresponding plasma [134]. IL-22 was also found to be abundant in both TB-induced pleural and pericardial effusions [135]. Therefore, it illustrated a possible aggregation of IL-22-producing cells in the disease sites of TB patients. MTBs in pulmonary TB granulomas have been shown to recruit IL-22+ T cells [136]. During MTB infection, since IL-22R is mainly expressed on the surfaces of macrophages in tuberculous granulomas, it may also contribute to Th22 cell accumulation [132,137][132][137]. Furthermore, in tuberculous pleurisy, the accumulation of Th22 cells at the disease site was associated with the chemotactic effect of cytokines in tuberculous pleural effusion (TPE) and the pleural mesothelial cell (PMC)-expressed chemokines CCL20, CCL22 and CCL27 [138]. PMCs also promote Th22 proliferation and differentiation by presenting MTB antigens [138]. MTB infection in humans can result in asymptomatic specific immune responses, latent tuberculosis (TB) or active TB [128]. Bunjun et al. observed a high level of IL-22 in latent MTB patients stimulated by MTB antigens, and IL-22 accounted for the largest proportion of responsive CD4+ responses [131]. Compared to wild-type controls, IL-22-/- mice showed higher susceptibility to MTB HN878 and a higher bacterial load in the lungs during the chronic phase of MTB infection [132]. In addition, the pulmonary MTB load at the early stage is significantly increased in IL-22-deficient mice [139]. Therefore, IL-22 is required in both adaptive and innate immune responses against MTB. Research has shown that in response to stimulation by MTB, Th22 cells can evolve into membrane-bound IL-22+ (mIL-22+) Th22 cells to extend the half-life of IL-22 [137]. More importantly, mIL-22 binds to IL-22R on infected macrophages to inhibit intracellular MTB replication [137]. IL-22 also enhances the expression of CCL2 on epithelial cells to stimulate the pulmonary recruitment of macrophages [132]. In MTB-infected phagocytes, IL-22 modulates the expression of Rab7 and Rab14 by upregulating the production of calgranulin A [140,141][140][141]. Rab7 and Rab14 subsequently inhibit intracellular MTB replication and promote phagosome maturation and fusion [140,141][140][141]. Furthermore, IL-22 was found to stimulate TNF-α production by IL-22R+ macrophages [132]. TNF-α can promote macrophage activation and MTB control directly. Additionally, the level of IL-22 in tuberculous pericardial effusion is positively correlated with MMP-9, an enzyme capable of degrading the extracellular matrix [135]. Since the peripheral level of MMP-9 is associated with the severity of tuberculosis [142], IL-22 might also regulate MMP-9 expression to control MTB infection [135]. Moreover, IL-22 can promote the proliferation and recovery of pleural mesothelial cells (PMCs) and the closure of PMC layers [138], which facilitates protection of PMCs from tuberculosis-induced damage. Antimicrobial proteins induced by IL-22 in the chronic phase, such as RegIIIγ, Lcn2 and calgranulin A, ensure the structural and functional integrity of the pulmonary epithelial barrier [132]. IL-22 is also involved in the formation of protective lymphoid follicular structures, called inducible bronchus-associated lymphoid tissues (iBALT), through the CXCR5–CXCL13 axis [139]. In patients infected with multidrug-resistant MTB (MDR-TB), decreased Th22 cell responses are associated with high sputum bacterial loads and severe lung lesions, suggesting that Th22 cells influence the antimicrobial capacity of TB patients [129]. Moreover, fewer T cells that release PD-1 or CD57 were found in multidrug-resistant TB patients with high levels of Th22 cells [129]. CD57 and programmed cell death 1 (PD-1) are markers of cellular senescence, so this result suggests that the Th22-induced anti-tuberculosis immune response may also be related to decreased T-cell senescence. Therefore, Th22 cells and IL-22 inhibit the progression of TB by protecting the pulmonary epithelial barrier and regulating MTB-specific immune responses.2.2.2. Citrobacter Rodentium
Since Citrobacter rodentium-infected mice closely resemble human infectious colitis, they are commonly used in colitis-associated scientific research [143]. C. rodentium infection in mice dramatically induces the expression of IL-22 in the intestinal mucosa [143]. Studies have confirmed that during C. rodentium infection, early IL-22 production is primarily dependent on ILC3s, whereas IL-22 derived from Th22 cells predominates at a later stage [144]. Th17 cells are also involved in IL-22 production [144]. Further experiments injected purified Th22 cells, Th17 cells or ILC3s into C. rodentium-infected mice deficient in IL-22. Compared to the other two subsets, mice injected with Th22 cells had a higher survival rate, reflecting that Th22/IL-22 is critical for the host’s immune response to C. rodentium [144]. Moreover, when the STAT3 gene was mutated, C. rodentium-infected mice showed a loss of Th22-induced immune defense, including impairment of the intestinal epithelial barrier, reduction of antimicrobial peptides, and increased dissemination of pathogenic bacteria [143]. This confirms that STAT3 is indispensable for the protective role of IL-22 during C. rodentium infection. In addition to STAT3, Th22/IL-22-related effects are dependent on T-bet and AhR [143,144][143][144]. Furthermore, IL-22 can induce colonic epithelial cells to express the antibacterial proteins RegIIIβ and RegIIIγ, both of which play a direct bactericidal role in C. rodentium infection [37]. Researchers have shown that antibiotic treatment reduces RegIIIγ production in mice, but oral administration of lipopolysaccharide (LPS) can restore it [145]. This indicates that LPS, the principal ingredient of the Gram-negative bacterial outer membrane, is key to the expression of RegIIIγ [145]. The addition of the Toll-like receptor (TLR)-5 agonist flagellin or the binding between TLR and its ligand MyD88 has been revealed to initiate the production of IL-23 by DCs and promote IL-23 to upregulate IL-22 production and RegIIIγ expression [146,147][146][147]. As a consequence, the host defense against C. rodentium induced by Th22 cells is dependent on STAT3 activation and antibacterial protein expression.2.2.3. Streptococcus pneumoniae
Streptococcus pneumoniae is a Gram-positive bacterium that colonizes the nasopharynx and initiates respiratory inflammation [148]. According to previous studies, IL-22 expression was rapidly upregulated in the lungs of mice suffering from pneumococcal pneumonia [149]. Moreover, the lung bacterial load was significantly higher in mice with hepatic IL-22R1 deficiency than in controls [149]. Hence, the IL-22-induced downstream cascade is critical for inhibiting S. pneumoniae replication. IL-22 was confirmed to promote the clearance of S. pneumoniae by upregulating complement factor C3’s expression in the liver and strengthening the binding of C3 and S. pneumoniae in the serum [149]. In addition, mice deficient in IL-22RA2 (the gene encoding IL-22BP) showed reduced susceptibility to S. pneumoniae and prolonged survival after infection [55]. Further analysis revealed that in the lungs of IL-22RA2-/- mice, oxidative phosphorylation-related genes were reduced in expression. Downregulated oxidative phosphorylation led to an increase in glycolysis and promoted a shift in macrophage phenotypes toward a proinflammatory phenotype, which ultimately upregulated host resistance to pneumococcal pneumonia. Consequently, IL-22 and IL-22BP coregulate antimicrobial immunity against S. pneumoniae, suggesting that IL-22RA2 may be an effective target for the treatment of pneumococcal pneumonia [55].2.2.4. Helicobacter pylori
Helicobacter pylori is Gram-negative and colonizes the gastric mucosa, causing persistent gastric inflammation, peptic ulcers or even gastric cancer [150,151][150][151]. Zhuang et al. demonstrated that Th22 cells are proinflammatory in H. pylori-triggered gastritis [152]. Upon H. pylori infection, IL-23-induced Th22 cells are rapidly enriched in the gastric mucosa and secrete IL-22, which enhances CXCL2 production by gastric epithelial cells [152]. CXCL2 subsequently bound to its corresponding receptors and resulted in the migration of myeloid-derived suppressor cells (MDSCs) toward the gastric epithelium. In response to IL-22 induction, MDSCs produce the proinflammatory factors calgranulin A (S100A8) and S100A9 and directly inhibit the development of Th1 cells, leading to gastritis progression [152]. Moreover, through activation of the ERK pathway, IL-22 and Helicobacter pylori synergistically promote the production of matrix metalloproteinases (MMPs), particularly MMP-10, in gastric epithelial cells [153]. MMP-10 can aggravate bacterial colonization by inhibiting the production of antimicrobial peptides in the gastric mucosa. In addition, MMP-10 also induces gastric epithelial cells to secrete the chemokine CXCL16, which recruits CD8+ T cells to the gastric mucosa and exacerbates the inflammatory response. However, when IL-22 acts synergistically with IL-17A, it stimulates antimicrobial peptide expression that protects the body from H. pylori infection [154]. Moreover, it has been proposed that due to the compensatory effect of other cytokines, IL-22 deficiency does not affect the susceptibility of mice to H. pylori or their mortality after infection [154], which contradicts the previous conclusion. According to a paper published in 2019, the discrepancy may be attributed to their choice of different mouse species and H. pylori strains, which closely influence the observed immune response [150]. Therefore, the role of IL-22/Th22 in H. pylori infection remains to be further discussed.2.2.5. Other Bacterial Infections
Clostridium difficile typically causes pseudomembranous colitis or bacterial diarrhea in immunocompromised individuals by facilitating the translocation of enteropathogenic bacteria [155,156][155][156]. Compared to the wild-type group, IL-22-deficient mice showed increased mortality after infection with C. difficile [156]. During C. difficile infection, induced IL-22 upregulates the expression of acute-phase proteins and C3 in the liver and intestine [156]. C3 can be deposited on the surface of enteropathogenic bacteria and enhance the bactericidal activity of neutrophils [156]. Moreover, IL-22-induced glycosylation of host N-linked glycans can promote the growth of Phascolarctobacterium spp. [157]. Phascolarctobacterium spp. is a succinate-consuming commensal bacterium that competes with C. difficile for energy, preventing the growth and colonization of C. difficile in the intestine. Infection with Klebsiella pneumoniae is often associated with healthcare-related pneumonia or sepsis [158]. Compared with uninfected mice, IL-22 levels in the lungs of mice infected with K. pneumoniae were upregulated [44]. The upregulation of IL-22 can reverse the decreased epithelial barrier stability and deteriorated pulmonary inflammation caused by IFN-λ [159]. IL-22 also induces the expression of defense genes such as lipocalin 2 to suppress K. pneumoniae [160]. Lipocalin 2 can sequester iron from bacteria to limit their growth [160,161][160][161]. Moreover, IL-22 stimulates the lung tissues of K. pneumoniae-infected mice to express CCL17 and CCL20 [44], which are ligands of CCR4 and CCR6 [17], leading to the accumulation of Th22 cells in the lungs. Pseudomonas aeruginosa is an opportunistic bacterium that can cause an overwhelming local immune response and consistently lead to acute respiratory distress syndrome (ARDS) [162]. The level of IL-22 was transiently increased in P. aeruginosa-infected mice, and additional administration of IL-22 reduced their pulmonary damage by downregulating local neutrophil infiltration [163]. IL-22 also induces pulmonary IFN-λ production, preventing the release of inflammatory mediators such as IL-1β [164]. Notably, both protease IV secreted by P. aeruginosa [165] and serine protease-3 secreted by neutrophils [166] can degrade IL-22. Therefore, they facilitate the immune escape of P. aeruginosa and can result in pulmonary bacterial colonization and the continued deterioration of respiratory function. Consequently, Th22/IL-22 is protective during C. difficile, K. pneumoniae and P. aeruginosa infections. However, Salmonella enterica serotype Typhimurium colonizes the gastrointestinal mucosa and typically causes inflammatory diarrhea [167]. The iroBCDE iroN gene cluster [168] and zinc transporter (ZnuABC) [169] in S. Typhimurium enable them to escape from IL-22-mediated metal chelation, called nutritional immunity [170]. Since IL-22 is primarily secreted by ILC3s rather than T cells during S. Typhimurium infection [171], more detailed mechanisms will not be discussed in this reviewere. Therefore, Th22/IL-22 exerts a crucial effect on regulating the immune response to viral and bacterial infections. Based on the above, cytokines such as TNF-α, IFN-λ, IL-10 and IL-23; transcription factors such as T-bet and AhR; and chemokines such as CCL2, CCL17 and CXCL13 have been shown to have a protective role in the Th22/IL-22 axis. Other effector molecules, including C3, CRP, MMP-9, Rab7, Rab14, the TLR-5 agonist flagellin and the antibacterial proteins RegIIIβ and RegIIIγ, are also involved in IL-22-induced anti-infectious immunity. Moreover, the matrix metalloproteinase MMP-10; the antimicrobial protein S100A9; and chemokines such as CXCL9, CXCL10, CXCL2 and CXCL16 are risk factors for the Th22/IL-22 downstream signaling system. Interestingly, STAT3 and the antimicrobial protein calgranulin A can be both protective and deleterious in the Th22/IL-22 axis, depending on the type of infectious disease. Notably, IL-22 in infectious illnesses is also derived from ILC3s, Th1 cells, Th17 cells and NKT cells, indicating that the interregulation between different cells ultimately contributes to the development of diseases. This suggests the need for more research on the underlying mechanisms of Th22/IL-22 in infectious diseases.References
- Ruterbusch, M.; Pruner, K.B.; Shehata, L.; Pepper, M. In Vivo CD4(+) T Cell Differentiation and Function: Revisiting the Th1/Th2 Paradigm. Annu. Rev. Immunol. 2020, 38, 705–725.
- Zhou, L.; Chong, M.M.; Littman, D.R. Plasticity of CD4+ T cell lineage differentiation. Immunity 2009, 30, 646–655.
- Xiao, F.; Han, M.; Rui, K.; Ai, X.; Tian, J.; Zhang, W.; Zhao, F.; Zhao, Y.; Jiang, Q.; Lu, L. New insights into follicular helper T cell response and regulation in autoimmune pathogenesis. Cell. Mol. Immunol. 2021, 18, 1610–1612.
- Wang, W.; Sung, N.; Gilman-Sachs, A.; Kwak-Kim, J. T Helper (Th) Cell Profiles in Pregnancy and Recurrent Pregnancy Losses: Th1/Th2/Th9/Th17/Th22/Tfh Cells. Front. Immunol. 2020, 11, 2025.
- Chatzileontiadou, D.S.M.; Sloane, H.; Nguyen, A.T.; Gras, S.; Grant, E.J. The Many Faces of CD4(+) T Cells: Immunological and Structural Characteristics. Int. J. Mol. Sci. 2020, 22, 73.
- Dumoutier, L.; Louahed, J.; Renauld, J.C. Cloning and characterization of IL-10-related T cell-derived inducible factor (IL-TIF), a novel cytokine structurally related to IL-10 and inducible by IL-9. J. Immunol. 2000, 164, 1814–1819.
- Wolk, K.; Kunz, S.; Asadullah, K.; Sabat, R. Cutting edge: Immune cells as sources and targets of the IL-10 family members? J. Immunol. 2002, 168, 5397–5402.
- Sheppard, P.; Kindsvogel, W.; Xu, W.; Henderson, K.; Schlutsmeyer, S.; Whitmore, T.E.; Kuestner, R.; Garrigues, U.; Birks, C.; Roraback, J.; et al. IL-28, IL-29 and their class II cytokine receptor IL-28R. Nat. Immunol. 2003, 4, 63–68.
- Gurney, A.L. IL-22, a Th1 cytokine that targets the pancreas and select other peripheral tissues. Int. Immunopharmacol. 2004, 4, 669–677.
- Wolk, K.; Sabat, R. Interleukin-22: A novel T- and NK-cell derived cytokine that regulates the biology of tissue cells. Cytokine Growth Factor Rev. 2006, 17, 367–380.
- Zheng, Y.; Danilenko, D.M.; Valdez, P.; Kasman, I.; Eastham-Anderson, J.; Wu, J.; Ouyang, W. Interleukin-22, a T(H)17 cytokine, mediates IL-23-induced dermal inflammation and acanthosis. Nature 2007, 445, 648–651.
- Kreymborg, K.; Etzensperger, R.; Dumoutier, L.; Haak, S.; Rebollo, A.; Buch, T.; Heppner, F.L.; Renauld, J.C.; Becher, B. IL-22 is expressed by Th17 cells in an IL-23-dependent fashion, but not required for the development of autoimmune encephalomyelitis. J. Immunol. 2007, 179, 8098–8104.
- Duhen, T.; Geiger, R.; Jarrossay, D.; Lanzavecchia, A.; Sallusto, F. Production of interleukin 22 but not interleukin 17 by a subset of human skin-homing memory T cells. Nat. Immunol. 2009, 10, 857–863.
- Trifari, S.; Kaplan, C.D.; Tran, E.H.; Crellin, N.K.; Spits, H. Identification of a human helper T cell population that has abundant production of interleukin 22 and is distinct from T(H)-17, T(H)1 and T(H)2 cells. Nat. Immunol. 2009, 10, 864–871.
- Eyerich, S.; Eyerich, K.; Pennino, D.; Carbone, T.; Nasorri, F.; Pallotta, S.; Cianfarani, F.; Odorisio, T.; Traidl-Hoffmann, C.; Behrendt, H.; et al. Th22 cells represent a distinct human T cell subset involved in epidermal immunity and remodeling. J. Clin. Investig. 2009, 119, 3573–3585.
- Dudakov, J.A.; Hanash, A.M.; van den Brink, M.R. Interleukin-22: Immunobiology and pathology. Annu. Rev. Immunol. 2015, 33, 747–785.
- Mousset, C.M.; Hobo, W.; Woestenenk, R.; Preijers, F.; Dolstra, H.; van der Waart, A.B. Comprehensive Phenotyping of T Cells Using Flow Cytometry. Cytometry. Part A J. Int. Soc. Anal. Cytol. 2019, 95, 647–654.
- Fujita, H.; Nograles, K.E.; Kikuchi, T.; Gonzalez, J.; Carucci, J.A.; Krueger, J.G. Human Langerhans cells induce distinct IL-22-producing CD4+ T cells lacking IL-17 production. Proc. Natl. Acad. Sci. USA 2009, 106, 21795–21800.
- Sommer, A.; Fabri, M. Vitamin D regulates cytokine patterns secreted by dendritic cells to promote differentiation of IL-22-producing T cells. PLoS ONE 2015, 10, e0130395.
- Lopez, D.V.; Al-Jaberi, F.A.H.; Damas, N.D.; Weinert, B.T.; Pus, U.; Torres-Rusillo, S.; Woetmann, A.; Ødum, N.; Bonefeld, C.M.; Kongsbak-Wismann, M.; et al. Vitamin D Inhibits IL-22 Production Through a Repressive Vitamin D Response Element in the il22 Promoter. Front. Immunol. 2021, 12, 715059.
- Huang, R.; Chen, X.; Long, Y.; Chen, R. MiR-31 promotes Th22 differentiation through targeting Bach2 in coronary heart disease. Biosci. Rep. 2019, 39, 986.
- Zeng, C.; Shao, Z.; Wei, Z.; Yao, J.; Wang, W.; Yin, L.; YangOu, H.; Xiong, D. The NOTCH-HES-1 axis is involved in promoting Th22 cell differentiation. Cell. Mol. Biol. Lett. 2021, 26, 7.
- Alam, M.S.; Maekawa, Y.; Kitamura, A.; Tanigaki, K.; Yoshimoto, T.; Kishihara, K.; Yasutomo, K. Notch signaling drives IL-22 secretion in CD4+ T cells by stimulating the aryl hydrocarbon receptor. Proc. Natl. Acad. Sci. USA. 2010, 107, 5943–5948.
- Niu, Y.; Ye, L.; Peng, W.; Wang, Z.; Wei, X.; Wang, X.; Li, Y.; Zhang, S.; Xiang, X.; Zhou, Q. IL-26 promotes the pathogenesis of malignant pleural effusion by enhancing CD4(+) IL-22(+) T-cell differentiation and inhibiting CD8(+) T-cell cytotoxicity. J. Leukoc. Biol. 2021, 110, 39–52.
- Fu, D.; Song, X.; Hu, H.; Sun, M.; Li, Z.; Tian, Z. Downregulation of RUNX3 moderates the frequency of Th17 and Th22 cells in patients with psoriasis. Mol. Med. Rep. 2016, 13, 4606–4612.
- Yang, J.; Yang, X.; Wang, L.; Li, M. B cells control lupus autoimmunity by inhibiting Th17 and promoting Th22 cells. Cell Death Dis. 2020, 11, 164.
- Yeste, A.; Mascanfroni, I.D.; Nadeau, M.; Burns, E.J.; Tukpah, A.M.; Santiago, A.; Wu, C.; Patel, B.; Kumar, D.; Quintana, F.J. IL-21 induces IL-22 production in CD4+ T cells. Nat. Commun. 2014, 5, 3753.
- Plank, M.W.; Kaiko, G.E.; Maltby, S.; Weaver, J.; Tay, H.L.; Shen, W.; Wilson, M.S.; Durum, S.K.; Foster, P.S. Th22 Cells Form a Distinct Th Lineage from Th17 Cells In Vitro with Unique Transcriptional Properties and Tbet-Dependent Th1 Plasticity. J. Immunol. 2017, 198, 2182–2190.
- Barnes, J.L.; Plank, M.W.; Asquith, K.; Maltby, S.; Sabino, L.R.; Kaiko, G.E.; Lochrin, A.; Horvat, J.C.; Mayall, J.R.; Kim, R.Y.; et al. T-helper 22 cells develop as a distinct lineage from Th17 cells during bacterial infection and phenotypic stability is regulated by T-bet. Mucosal Immunol. 2021, 14, 1077–1087.
- Ouyang, W.; O’Garra, A. IL-10 Family Cytokines IL-10 and IL-22: From Basic Science to Clinical Translation. Immunity 2019, 50, 871–891.
- Logsdon, N.J.; Jones, B.C.; Josephson, K.; Cook, J.; Walter, M.R. Comparison of interleukin-22 and interleukin-10 soluble receptor complexes. J. Interferon Cytokine Res. Off. J. Int. Soc. Interferon Cytokine Res. 2002, 22, 1099–1112.
- Li, J.; Tomkinson, K.N.; Tan, X.Y.; Wu, P.; Yan, G.; Spaulding, V.; Deng, B.; Annis-Freeman, B.; Heveron, K.; Zollner, R.; et al. Temporal associations between interleukin 22 and the extracellular domains of IL-22R and IL-10R2. Int. Immunopharmacol. 2004, 4, 693–708.
- Bleicher, L.; de Moura, P.R.; Watanabe, L.; Colau, D.; Dumoutier, L.; Renauld, J.C.; Polikarpov, I. Crystal structure of the IL-22/IL-22R1 complex and its implications for the IL-22 signaling mechanism. FEBS Lett. 2008, 582, 2985–2992.
- Wolk, K.; Kunz, S.; Witte, E.; Friedrich, M.; Asadullah, K.; Sabat, R. IL-22 increases the innate immunity of tissues. Immunity 2004, 21, 241–254.
- Jiang, Q.; Yang, G.; Xiao, F.; Xie, J.; Wang, S.; Lu, L.; Cui, D. Role of Th22 Cells in the Pathogenesis of Autoimmune Diseases. Front. Immunol. 2021, 12, 688066.
- Ahn, D.; Prince, A. Participation of the IL-10RB Related Cytokines, IL-22 and IFN-λ in Defense of the Airway Mucosal Barrier. Front. Cell. Infect. Microbiol. 2020, 10, 300.
- Zheng, Y.; Valdez, P.A.; Danilenko, D.M.; Hu, Y.; Sa, S.M.; Gong, Q.; Abbas, A.R.; Modrusan, Z.; Ghilardi, N.; de Sauvage, F.J.; et al. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat. Med. 2008, 14, 282–289.
- Wolk, K.; Witte, E.; Wallace, E.; Döcke, W.D.; Kunz, S.; Asadullah, K.; Volk, H.D.; Sterry, W.; Sabat, R. IL-22 regulates the expression of genes responsible for antimicrobial defense, cellular differentiation, and mobility in keratinocytes: A potential role in psoriasis. Eur. J. Immunol. 2006, 36, 1309–1323.
- Boniface, K.; Bernard, F.X.; Garcia, M.; Gurney, A.L.; Lecron, J.C.; Morel, F. IL-22 inhibits epidermal differentiation and induces proinflammatory gene expression and migration of human keratinocytes. J. Immunol. 2005, 174, 3695–3702.
- Wolk, K.; Haugen, H.S.; Xu, W.; Witte, E.; Waggie, K.; Anderson, M.; Vom Baur, E.; Witte, K.; Warszawska, K.; Philipp, S.; et al. IL-22 and IL-20 are key mediators of the epidermal alterations in psoriasis while IL-17 and IFN-gamma are not. J. Mol. Med. Berl.Ger. 2009, 87, 523–536.
- Hebert, K.D.; McLaughlin, N.; Galeas-Pena, M.; Zhang, Z.; Eddens, T.; Govero, A.; Pilewski, J.M.; Kolls, J.K.; Pociask, D.A. Targeting the IL-22/IL-22BP axis enhances tight junctions and reduces inflammation during influenza infection. Mucosal Immunol. 2020, 13, 64–74.
- Pickert, G.; Neufert, C.; Leppkes, M.; Zheng, Y.; Wittkopf, N.; Warntjen, M.; Lehr, H.A.; Hirth, S.; Weigmann, B.; Wirtz, S.; et al. STAT3 links IL-22 signaling in intestinal epithelial cells to mucosal wound healing. J. Exp. Med. 2009, 206, 1465–1472.
- Sonnenberg, G.F.; Nair, M.G.; Kirn, T.J.; Zaph, C.; Fouser, L.A.; Artis, D. Pathological versus protective functions of IL-22 in airway inflammation are regulated by IL-17A. J. Exp. Med. 2010, 207, 1293–1305.
- Aujla, S.J.; Chan, Y.R.; Zheng, M.; Fei, M.; Askew, D.J.; Pociask, D.A.; Reinhart, T.A.; McAllister, F.; Edeal, J.; Gaus, K.; et al. IL-22 mediates mucosal host defense against Gram-negative bacterial pneumonia. Nat. Med. 2008, 14, 275–281.
- Mitra, A.; Raychaudhuri, S.K.; Raychaudhuri, S.P. IL-22 induced cell proliferation is regulated by PI3K/Akt/mTOR signaling cascade. Cytokine 2012, 60, 38–42.
- Li, H.; Zhang, Q.; Wu, Q.; Cui, Y.; Zhu, H.; Fang, M.; Zhou, X.; Sun, Z.; Yu, J. Interleukin-22 secreted by cancer-associated fibroblasts regulates the proliferation and metastasis of lung cancer cells via the PI3K-Akt-mTOR signaling pathway. Am. J. Transl. Res. 2019, 11, 4077–4088.
- Nagalakshmi, M.L.; Rascle, A.; Zurawski, S.; Menon, S.; de Waal Malefyt, R. Interleukin-22 activates STAT3 and induces IL-10 by colon epithelial cells. Int. Immunopharmacol. 2004, 4, 679–691.
- Resham, S.; Saalim, M.; Manzoor, S.; Ahmad, H.; Bangash, T.A.; Latif, A.; Jaleel, S. Mechanistic study of interaction between IL-22 and HCV core protein in the development of hepatocellular carcinoma among liver transplant recipients. Microb. Pathog. 2020, 142, 104071.
- Dumoutier, L.; Van Roost, E.; Colau, D.; Renauld, J.C. Human interleukin-10-related T cell-derived inducible factor: Molecular cloning and functional characterization as an hepatocyte-stimulating factor. Proc. Natl. Acad. Sci. USA 2000, 97, 10144–10149.
- Quiñones-Mateu, M.E.; Lederman, M.M.; Feng, Z.; Chakraborty, B.; Weber, J.; Rangel, H.R.; Marotta, M.L.; Mirza, M.; Jiang, B.; Kiser, P.; et al. Human epithelial beta-defensins 2 and 3 inhibit HIV-1 replication. AIDS Lond. Engl. 2003, 17, F39–F48.
- Sertorio, M.; Hou, X.; Carmo, R.F.; Dessein, H.; Cabantous, S.; Abdelwahed, M.; Romano, A.; Albuquerque, F.; Vasconcelos, L.; Carmo, T.; et al. IL-22 and IL-22 binding protein (IL-22BP) regulate fibrosis and cirrhosis in hepatitis C virus and schistosome infections. Hepatol. Baltim. Md. 2015, 61, 1321–1331.
- Dumoutier, L.; Lejeune, D.; Colau, D.; Renauld, J.C. Cloning and characterization of IL-22 binding protein, a natural antagonist of IL-10-related T cell-derived inducible factor/IL-22. J. Immunol. 2001, 166, 7090–7095.
- Xu, W.; Presnell, S.R.; Parrish-Novak, J.; Kindsvogel, W.; Jaspers, S.; Chen, Z.; Dillon, S.R.; Gao, Z.; Gilbert, T.; Madden, K.; et al. A soluble class II cytokine receptor, IL-22RA2, is a naturally occurring IL-22 antagonist. Proc. Natl. Acad. Sci. USA 2001, 98, 9511–9516.
- Abood, R.N.; McHugh, K.J.; Rich, H.E.; Ortiz, M.A.; Tobin, J.M.; Ramanan, K.; Robinson, K.M.; Bomberger, J.M.; Kolls, J.K.; Manni, M.L.; et al. IL-22-binding protein exacerbates influenza, bacterial super-infection. Mucosal Immunol. 2019, 12, 1231–1243.
- Trevejo-Nunez, G.; Elsegeiny, W.; Aggor, F.E.Y.; Tweedle, J.L.; Kaplan, Z.; Gandhi, P.; Castillo, P.; Ferguson, A.; Alcorn, J.F.; Chen, K.; et al. Interleukin-22 (IL-22) Binding Protein Constrains IL-22 Activity, Host Defense, and Oxidative Phosphorylation Genes during Pneumococcal Pneumonia. Infect. Immun. 2019, 87, e00550-19.
- Hoffmann, J.P.; Kolls, J.K.; McCombs, J.E. Regulation and Function of ILC3s in Pulmonary Infections. Front. Immunol. 2021, 12, 672523.
- Pociask, D.A.; Scheller, E.V.; Mandalapu, S.; McHugh, K.J.; Enelow, R.I.; Fattman, C.L.; Kolls, J.K.; Alcorn, J.F. IL-22 is essential for lung epithelial repair following influenza infection. Am. J. Pathol. 2013, 182, 1286–1296.
- Das, S.; St Croix, C.; Good, M.; Chen, J.; Zhao, J.; Hu, S.; Ross, M.; Myerburg, M.M.; Pilewski, J.M.; Williams, J.; et al. Interleukin-22 Inhibits Respiratory Syncytial Virus Production by Blocking Virus-Mediated Subversion of Cellular Autophagy. iScience 2020, 23, 101256.
- Barhoum, P.; Pineton de Chambrun, M.; Dorgham, K.; Kerneis, M.; Burrel, S.; Quentric, P.; Parizot, C.; Chommeloux, J.; Bréchot, N.; Moyon, Q.; et al. Phenotypic Heterogeneity of Fulminant COVID-19--Related Myocarditis in Adults. J. Am. Coll. Cardiol. 2022, 80, 299–312.
- Albayrak, N.; Orte Cano, C.; Karimi, S.; Dogahe, D.; Van Praet, A.; Godefroid, A.; Del Marmol, V.; Grimaldi, D.; Bondue, B.; Van Vooren, J.P.; et al. Distinct Expression Patterns of Interleukin-22 Receptor 1 on Blood Hematopoietic Cells in SARS-CoV-2 Infection. Front. Immunol. 2022, 13, 769839.
- Ahmed Mostafa, G.; Mohamed Ibrahim, H.; Al Sayed Shehab, A.; Mohamed Magdy, S.; AboAbdoun Soliman, N.; Fathy El-Sherif, D. Up-regulated serum levels of interleukin (IL)-17A and IL-22 in Egyptian pediatric patients with COVID-19 and MIS-C: Relation to the disease outcome. Cytokine 2022, 154, 155870.
- Ellison-Hughes, G.M.; Colley, L.; O’Brien, K.A.; Roberts, K.A.; Agbaedeng, T.A.; Ross, M.D. The Role of MSC Therapy in Attenuating the Damaging Effects of the Cytokine Storm Induced by COVID-19 on the Heart and Cardiovascular System. Front. Cardiovasc. Med. 2020, 7, 602183.
- Guzik, T.J.; Mohiddin, S.A.; Dimarco, A.; Patel, V.; Savvatis, K.; Marelli-Berg, F.M.; Madhur, M.S.; Tomaszewski, M.; Maffia, P.; D’Acquisto, F.; et al. COVID-19 and the cardiovascular system: Implications for risk assessment, diagnosis, and treatment options. Cardiovasc. Res. 2020, 116, 1666–1687.
- Shimabukuro-Vornhagen, A.; Gödel, P.; Subklewe, M.; Stemmler, H.J.; Schlößer, H.A.; Schlaak, M.; Kochanek, M.; Böll, B.; von Bergwelt-Baildon, M.S. Cytokine release syndrome. J. Immunother. Cancer 2018, 6, 56.
- Lee, D.W.; Gardner, R.; Porter, D.L.; Louis, C.U.; Ahmed, N.; Jensen, M.; Grupp, S.A.; Mackall, C.L. Current concepts in the diagnosis and management of cytokine release syndrome. Blood 2014, 124, 188–195.
- Fanales-Belasio, E.; Raimondo, M.; Suligoi, B.; Buttò, S. HIV virology and pathogenetic mechanisms of infection: A brief overview. Ann. Dell’istituto Super. Di Sanita 2010, 46, 5–14.
- Missé, D.; Yssel, H.; Trabattoni, D.; Oblet, C.; Lo Caputo, S.; Mazzotta, F.; Pène, J.; Gonzalez, J.P.; Clerici, M.; Veas, F. IL-22 participates in an innate anti-HIV-1 host-resistance network through acute-phase protein induction. J. Immunol. 2007, 178, 407–415.
- Morou, A.; Brunet-Ratnasingham, E.; Dubé, M.; Charlebois, R.; Mercier, E.; Darko, S.; Brassard, N.; Nganou-Makamdop, K.; Arumugam, S.; Gendron-Lepage, G.; et al. Altered differentiation is central to HIV-specific CD4(+) T cell dysfunction in progressive disease. Nat. Immunol. 2019, 20, 1059–1070.
- Uhlar, C.M.; Burgess, C.J.; Sharp, P.M.; Whitehead, A.S. Evolution of the serum amyloid A (SAA) protein superfamily. Genomics 1994, 19, 228–235.
- McKinnon, L.R.; Kaul, R. Quality and quantity: Mucosal CD4+ T cells and HIV susceptibility. Curr. Opin. HIV AIDS 2012, 7, 195–202.
- Veazey, R.S. Intestinal CD4 Depletion in HIV / SIV Infection. Curr. Immunol. Rev. 2019, 15, 76–91.
- Ryan, E.S.; Micci, L.; Fromentin, R.; Paganini, S.; McGary, C.S.; Easley, K.; Chomont, N.; Paiardini, M. Loss of Function of Intestinal IL-17 and IL-22 Producing Cells Contributes to Inflammation and Viral Persistence in SIV-Infected Rhesus Macaques. PLoS Pathog. 2016, 12, e1005412.
- Sonnenberg, G.F.; Monticelli, L.A.; Alenghat, T.; Fung, T.C.; Hutnick, N.A.; Kunisawa, J.; Shibata, N.; Grunberg, S.; Sinha, R.; Zahm, A.M.; et al. Innate lymphoid cells promote anatomical containment of lymphoid-resident commensal bacteria. Science 2012, 336, 1321–1325.
- Arias, J.F.; Nishihara, R.; Bala, M.; Ikuta, K. High systemic levels of interleukin-10, interleukin-22 and C-reactive protein in Indian patients are associated with low in vitro replication of HIV-1 subtype C viruses. Retrovirology 2010, 7, 15.
- Campillo-Gimenez, L.; Casulli, S.; Dudoit, Y.; Seang, S.; Carcelain, G.; Lambert-Niclot, S.; Appay, V.; Autran, B.; Tubiana, R.; Elbim, C. Neutrophils in antiretroviral therapy-controlled HIV demonstrate hyperactivation associated with a specific IL-17/IL-22 environment. J. Allergy Clin. Immunol. 2014, 134, 1142–1152.
- Fernandes, S.M.; Pires, A.R.; Matoso, P.; Ferreira, C.; Nunes-Cabaço, H.; Correia, L.; Valadas, E.; Poças, J.; Pacheco, P.; Veiga-Fernandes, H.; et al. HIV-2 infection is associated with preserved GALT homeostasis and epithelial integrity despite ongoing mucosal viral replication. Mucosal Immunol. 2018, 11, 236–248.
- Khaitan, A.; Kilberg, M.; Kravietz, A.; Ilmet, T.; Tastan, C.; Mwamzuka, M.; Marshed, F.; Liu, M.; Ahmed, A.; Borkowsky, W.; et al. HIV-Infected Children Have Lower Frequencies of CD8+ Mucosal-Associated Invariant T (MAIT) Cells that Correlate with Innate, Th17 and Th22 Cell Subsets. PLoS ONE 2016, 11, e0161786.
- Le Bourhis, L.; Martin, E.; Péguillet, I.; Guihot, A.; Froux, N.; Coré, M.; Lévy, E.; Dusseaux, M.; Meyssonnier, V.; Premel, V.; et al. Antimicrobial activity of mucosal-associated invariant T cells. Nat. Immunol. 2010, 11, 701–708.
- Ussher, J.E.; Klenerman, P.; Willberg, C.B. Mucosal-associated invariant T-cells: New players in anti-bacterial immunity. Front. Immunol. 2014, 5, 450.
- Meyer-Myklestad, M.H.; Medhus, A.W.; Lorvik, K.B.; Seljeflot, I.; Hansen, S.H.; Holm, K.; Stiksrud, B.; Trøseid, M.; Hov, J.R.; Kvale, D.; et al. Human Immunodeficiency Virus-Infected Immunological Nonresponders Have Colon-Restricted Gut Mucosal Immune Dysfunction. J. Infect. Dis. 2022, 225, 661–674.
- Lok, A.S.F.; McMahon, B.J. Chronic hepatitis B. N. Engl. J. Med. 2002, 346, 1682–1683.
- Manns, M.P.; McHutchison, J.G.; Gordon, S.C.; Rustgi, V.K.; Shiffman, M.; Reindollar, R.; Goodman, Z.D.; Koury, K.; Ling, M.; Albrecht, J.K. Peginterferon alfa-2b plus ribavirin compared with interferon alfa-2b plus ribavirin for initial treatment of chronic hepatitis C: A randomised trial. Lancet 2001, 358, 958–965.
- Lauer, G.M.; Walker, B.D. Hepatitis C virus infection. N. Engl. J. Med. 2001, 345, 41–52.
- Dambacher, J.; Beigel, F.; Zitzmann, K.; Heeg, M.H.; Göke, B.; Diepolder, H.M.; Auernhammer, C.J.; Brand, S. The role of interleukin-22 in hepatitis C virus infection. Cytokine 2008, 41, 209–216.
- Zenewicz, L.A.; Yancopoulos, G.D.; Valenzuela, D.M.; Murphy, A.J.; Karow, M.; Flavell, R.A. Interleukin-22 but not interleukin-17 provides protection to hepatocytes during acute liver inflammation. Immunity 2007, 27, 647–659.
- Feng, D.; Kong, X.; Weng, H.; Park, O.; Wang, H.; Dooley, S.; Gershwin, M.E.; Gao, B. Interleukin-22 promotes proliferation of liver stem/progenitor cells in mice and patients with chronic hepatitis B virus infection. Gastroenterology 2012, 143, 188–198.
- Zheng, W.P.; Zhang, B.Y.; Shen, Z.Y.; Yin, M.L.; Cao, Y.; Song, H.L. Biological effects of bone marrow mesenchymal stem cells on hepatitis B virus in vitro. Mol. Med. Rep. 2017, 15, 2551–2559.
- Zhao, J.; Zhang, Z.; Luan, Y.; Zou, Z.; Sun, Y.; Li, Y.; Jin, L.; Zhou, C.; Fu, J.; Gao, B.; et al. Pathological functions of interleukin-22 in chronic liver inflammation and fibrosis with hepatitis B virus infection by promoting T helper 17 cell recruitment. Hepatology 2014, 59, 1331–1342.
- Mo, R.; Wang, P.; Lai, R.; Li, F.; Liu, Y.; Jiang, S.; Zhao, G.; Guo, S.; Zhou, H.; Lin, L.; et al. Persistently elevated circulating Th22 reversely correlates with prognosis in HBV-related acute-on-chronic liver failure. J. Gastroenterol. Hepatol. 2017, 32, 677–686.
- Kong, X.; Feng, D.; Wang, H.; Hong, F.; Bertola, A.; Wang, F.S.; Gao, B. Interleukin-22 induces hepatic stellate cell senescence and restricts liver fibrosis in mice. Hepatology 2012, 56, 1150–1159.
- Xiang, X.; Gui, H.; King, N.J.; Cole, L.; Wang, H.; Xie, Q.; Bao, S. IL-22 and non-ELR-CXC chemokine expression in chronic hepatitis B virus-infected liver. Immunol. Cell Biol. 2012, 90, 611–619.
- Gao, W.; Fan, Y.C.; Zhang, J.Y.; Zheng, M.H. Emerging Role of Interleukin 22 in Hepatitis B Virus Infection: A Double-edged Sword. J. Clin. Transl. Hepatol. 2013, 1, 103–108.
- Cobleigh, M.A.; Robek, M.D. Protective and pathological properties of IL-22 in liver disease: Implications for viral hepatitis. Am. J. Pathol. 2013, 182, 21–28.
- Foster, R.G.; Golden-Mason, L.; Rutebemberwa, A.; Rosen, H.R. Interleukin (IL)-17/IL-22-producing T cells enriched within the liver of patients with chronic hepatitis C viral (HCV) infection. Dig. Dis. Sci. 2012, 57, 381–389.
- Wu, L.Y.; Liu, S.; Liu, Y.; Guo, C.; Li, H.; Li, W.; Jin, X.; Zhang, K.; Zhao, P.; Wei, L.; et al. Up-regulation of interleukin-22 mediates liver fibrosis via activating hepatic stellate cells in patients with hepatitis C. Clin. Immunol. 2015, 158, 77–87.
- Kong, F.; Zhang, W.; Feng, B.; Zhang, H.; Rao, H.; Wang, J.; Cong, X.; Wei, L. Abnormal CD4 + T helper (Th) 1 cells and activated memory B cells are associated with type III asymptomatic mixed cryoglobulinemia in HCV infection. Virol. J. 2015, 12, 100.
- De Brito, R.; do Carmo, R.F.; Silva, B.M.S.; Costa, A.C.S.; Rocha, S.W.S.; Vasconcelos, L.R.S.; Pereira, L.; de Moura, P. Liver expression of IL-22, IL-22R1 and IL-22BP in patients with chronic hepatitis C with different fibrosis stages. Cytokine 2022, 150, 155784.
- Zenewicz, L.A. IL-22 Binding Protein (IL-22BP) in the Regulation of IL-22 Biology. Front. Immunol. 2021, 12, 766586.
- Voglis, S.; Moos, S.; Kloos, L.; Wanke, F.; Zayoud, M.; Pelczar, P.; Giannou, A.D.; Pezer, S.; Albers, M.; Luessi, F.; et al. Regulation of IL-22BP in psoriasis. Sci. Rep. 2018, 8, 5085.
- Huber, S.; Gagliani, N.; Zenewicz, L.A.; Huber, F.J.; Bosurgi, L.; Hu, B.; Hedl, M.; Zhang, W.; O’Connor, W.; Murphy, A.J.; et al. IL-22BP is regulated by the inflammasome and modulates tumorigenesis in the intestine. Nature 2012, 491, 259–263.
- Wu, L.; Zhao, J. Does IL-22 protect against liver fibrosis in hepatitis C virus infection? Hepatology 2015, 62, 1919.
- Hebert, K.D.; McLaughlin, N.; Zhang, Z.; Cipriani, A.; Alcorn, J.F.; Pociask, D.A. IL-22Ra1 is induced during influenza infection by direct and indirect TLR3 induction of STAT1. Respir. Res. 2019, 20, 184.
- Guo, H.; Topham, D.J. Interleukin-22 (IL-22) production by pulmonary Natural Killer cells and the potential role of IL-22 during primary influenza virus infection. J. Virol. 2010, 84, 7750–7759.
- Paget, C.; Ivanov, S.; Fontaine, J.; Renneson, J.; Blanc, F.; Pichavant, M.; Dumoutier, L.; Ryffel, B.; Renauld, J.C.; Gosset, P.; et al. Interleukin-22 is produced by invariant natural killer T lymphocytes during influenza A virus infection: Potential role in protection against lung epithelial damages. J. Biol. Chem. 2012, 287, 8816–8829.
- Kumar, P.; Thakar, M.S.; Ouyang, W.; Malarkannan, S. IL-22 from conventional NK cells is epithelial regenerative and inflammation protective during influenza infection. Mucosal Immunol. 2013, 6, 69–82.
- Kudva, A.; Scheller, E.V.; Robinson, K.M.; Crowe, C.R.; Choi, S.M.; Slight, S.R.; Khader, S.A.; Dubin, P.J.; Enelow, R.I.; Kolls, J.K.; et al. Influenza A inhibits Th17-mediated host defense against bacterial pneumonia in mice. J. Immunol. 2011, 186, 1666–1674.
- Barthelemy, A.; Sencio, V.; Soulard, D.; Deruyter, L.; Faveeuw, C.; Le Goffic, R.; Trottein, F. Interleukin-22 Immunotherapy during Severe Influenza Enhances Lung Tissue Integrity and Reduces Secondary Bacterial Systemic Invasion. Infect. Immun. 2018, 86, e00706-17.
- Kaarteenaho, R.; Merikallio, H.; Lehtonen, S.; Harju, T.; Soini, Y. Divergent expression of claudin -1, -3, -4, -5 and -7 in developing human lung. Respir. Res. 2010, 11, 59.
- Eaton, D.C.; Helms, M.N.; Koval, M.; Bao, H.F.; Jain, L. The contribution of epithelial sodium channels to alveolar function in health and disease. Annu. Rev. Physiol. 2009, 71, 403–423.
- Ivanov, S.; Renneson, J.; Fontaine, J.; Barthelemy, A.; Paget, C.; Fernandez, E.M.; Blanc, F.; De Trez, C.; Van Maele, L.; Dumoutier, L.; et al. Interleukin-22 reduces lung inflammation during influenza A virus infection and protects against secondary bacterial infection. J. Virol. 2013, 87, 6911–6924.
- Xie, Y.; Yu, Y.; Zhao, L.; Ning, P.; Luo, Q.; Zhang, Y.; Yin, L.; Zheng, Y.; Gao, Z. Specific Cytokine Profiles Predict the Severity of Influenza A Pneumonia: A Prospectively Multicenter Pilot Study. BioMed Res. Int. 2021, 2021, 9533044.
- Crocker, S.J.; Frausto, R.F.; Whitmire, J.K.; Benning, N.; Milner, R.; Whitton, J.L. Amelioration of coxsackievirus B3-mediated myocarditis by inhibition of tissue inhibitors of matrix metalloproteinase-1. Am. J. Pathol. 2007, 171, 1762–1773.
- Kong, Q.; Wu, W.; Yang, F.; Liu, Y.; Xue, Y.; Gao, M.; Lai, W.; Pan, X.; Yan, Y.; Pang, Y.; et al. Increased expressions of IL-22 and Th22 cells in the coxsackievirus B3-Induced mice acute viral myocarditis. Virol. J. 2012, 9, 232.
- Guo, Y.; Wu, W.; Cen, Z.; Li, X.; Kong, Q.; Zhou, Q. IL-22-producing Th22 cells play a protective role in CVB3-induced chronic myocarditis and dilated cardiomyopathy by inhibiting myocardial fibrosis. Virol. J. 2014, 11, 230.
- Kong, Q.; Xue, Y.; Wu, W.; Yang, F.; Liu, Y.; Gao, M.; Lai, W.; Pan, X. IL-22 exacerbates the severity of CVB3-induced acute viral myocarditis in IL-17A-deficient mice. Mol. Med. Rep. 2013, 7, 1329–1335.
- Chan, K.P.; Goh, K.T.; Chong, C.Y.; Teo, E.S.; Lau, G.; Ling, A.E. Epidemic hand, foot and mouth disease caused by human enterovirus 71, Singapore. Emerg. Infect. Dis. 2003, 9, 78–85.
- Zhang, S.Y.; Xu, M.Y.; Xu, H.M.; Li, X.J.; Ding, S.J.; Wang, X.J.; Li, T.Y.; Lu, Q.B. Immunologic Characterization of Cytokine Responses to Enterovirus 71 and Coxsackievirus A16 Infection in Children. Medicine 2015, 94, e1137.
- Cui, D.; Zhong, F.; Lin, J.; Wu, Y.; Long, Q.; Yang, X.; Zhu, Q.; Huang, L.; Mao, Q.; Huo, Z.; et al. Changes of circulating Th22 cells in children with hand, foot, and mouth disease caused by enterovirus 71 infection. Oncotarget 2017, 8, 29370–29382.
- Hall, C.B. Respiratory syncytial virus and parainfluenza virus. N. Engl. J. Med. 2001, 344, 1917–1928.
- Nair, H.; Nokes, D.J.; Gessner, B.D.; Dherani, M.; Madhi, S.A.; Singleton, R.J.; O’Brien, K.L.; Roca, A.; Wright, P.F.; Bruce, N.; et al. Global burden of acute lower respiratory infections due to respiratory syncytial virus in young children: A systematic review and meta-analysis. Lancet 2010, 375, 1545–1555.
- Widmer, K.; Zhu, Y.; Williams, J.V.; Griffin, M.R.; Edwards, K.M.; Talbot, H.K. Rates of hospitalizations for respiratory syncytial virus, human metapneumovirus, and influenza virus in older adults. J. Infect. Dis. 2012, 206, 56–62.
- Geevarghese, B.; Weinberg, A. Cell-mediated immune responses to respiratory syncytial virus infection: Magnitude, kinetics, and correlates with morbidity and age. Hum. Vaccines Immunother. 2014, 10, 1047–1056.
- Soenjoyo, K.R.; Chua, B.W.B.; Wee, L.W.Y.; Koh, M.J.A.; Ang, S.B. Treatment of cutaneous viral warts in children: A review. Dermatol. Ther. 2020, 33, e14034.
- Marie, R.E.M.; Abuzeid, A.; Attia, F.M.; Anani, M.M.; Gomaa, A.H.A.; Atef, L.M. Serum level of interleukin-22 in patients with cutaneous warts: A case-control study. J. Cosmet. Dermatol. 2021, 20, 1782–1787.
- Ferreira, M.S.; Júnior, P.S.B.; Cerqueira, V.D.; Rivero, G.R.C.; Júnior, C.A.O.; Castro, P.H.G.; Silva, G.A.D.; Silva, W.B.D.; Imbeloni, A.A.; Sousa, J.R.; et al. Experimental yellow fever virus infection in the squirrel monkey (Saimiri spp.) I: Gross anatomical and histopathological findings in organs at necropsy. Mem. Do Inst. Oswaldo Cruz 2020, 115, e190501.
- De Rodaniche, E.; Galindo, P. Isolation of yellow fever virus from Haemagogus mesodentatus, H. equinus and Sabethes chloropterus captured in Guatemala in 1956. Am. J. Trop. Med. Hyg. 1957, 6, 232–237.
- Mendes, C.C.H.; de Sousa, J.R.; Olímpio, F.A.; Falcão, L.F.M.; Carvalho, M.L.G.; da Costa Lopes, J.; Martins Filho, A.J.; do Socorro Cabral Miranda, V.; Dos Santos, L.C.; da Silva Vilacoert, F.S.; et al. Th22 cytokines and yellow fever: Possible implications for the immunopathogenesis of human liver infection. Cytokine 2022, 157, 155924.
- Flynn, J.L.; Chan, J. Immunology of tuberculosis. Annu. Rev. Immunol. 2001, 19, 93–129.
- Imperiale, B.R.; García, A.; Minotti, A.; González Montaner, P.; Moracho, L.; Morcillo, N.S.; Palmero, D.J.; Sasiain, M.D.C.; de la Barrera, S. Th22 response induced by Mycobacterium tuberculosis strains is closely related to severity of pulmonary lesions and bacillary load in patients with multi-drug-resistant tuberculosis. Clin. Exp. Immunol. 2021, 203, 267–280.
- Cowan, J.; Pandey, S.; Filion, L.G.; Angel, J.B.; Kumar, A.; Cameron, D.W. Comparison of interferon-γ-, interleukin (IL)-17- and IL-22-expressing CD4 T cells, IL-22-expressing granulocytes and proinflammatory cytokines during latent and active tuberculosis infection. Clin. Exp. Immunol. 2012, 167, 317–329.
- Bunjun, R.; Omondi, F.M.A.; Makatsa, M.S.; Keeton, R.; Wendoh, J.M.; Müller, T.L.; Prentice, C.S.L.; Wilkinson, R.J.; Riou, C.; Burgers, W.A. Th22 Cells Are a Major Contributor to the Mycobacterial CD4(+) T Cell Response and Are Depleted During HIV Infection. J. Immunol. 2021, 207, 1239–1249.
- Treerat, P.; Prince, O.; Cruz-Lagunas, A.; Muñoz-Torrico, M.; Salazar-Lezama, M.A.; Selman, M.; Fallert-Junecko, B.; Reinhardt, T.A.; Alcorn, J.F.; Kaushal, D.; et al. Novel role for IL-22 in protection during chronic Mycobacterium tuberculosis HN878 infection. Mucosal Immunol. 2017, 10, 1069–1081.
- Behrends, J.; Renauld, J.C.; Ehlers, S.; Hölscher, C. IL-22 is mainly produced by IFNγ-secreting cells but is dispensable for host protection against Mycobacterium tuberculosis infection. PLoS ONE 2013, 8, e57379.
- Scriba, T.J.; Kalsdorf, B.; Abrahams, D.A.; Isaacs, F.; Hofmeister, J.; Black, G.; Hassan, H.Y.; Wilkinson, R.J.; Walzl, G.; Gelderbloem, S.J.; et al. Distinct, specific IL-17- and IL-22-producing CD4+ T cell subsets contribute to the human anti-mycobacterial immune response. J. Immunol. 2008, 180, 1962–1970.
- Matthews, K.; Wilkinson, K.A.; Kalsdorf, B.; Roberts, T.; Diacon, A.; Walzl, G.; Wolske, J.; Ntsekhe, M.; Syed, F.; Russell, J.; et al. Predominance of interleukin-22 over interleukin-17 at the site of disease in human tuberculosis. Tuberc. Edinb. Scotl. 2011, 91, 587–593.
- Yao, S.; Huang, D.; Chen, C.Y.; Halliday, L.; Zeng, G.; Wang, R.C.; Chen, Z.W. Differentiation, distribution and gammadelta T cell-driven regulation of IL-22-producing T cells in tuberculosis. PLoS Pathog. 2010, 6, e1000789.
- Zeng, G.; Chen, C.Y.; Huang, D.; Yao, S.; Wang, R.C.; Chen, Z.W. Membrane-bound IL-22 after de novo production in tuberculosis and anti-Mycobacterium tuberculosis effector function of IL-22+ CD4+ T cells. J. Immunol. 2011, 187, 190–199.
- Ye, Z.J.; Zhou, Q.; Yuan, M.L.; Du, R.H.; Yang, W.B.; Xiong, X.Z.; Huang, B.; Shi, H.Z. Differentiation and recruitment of IL-22-producing helper T cells stimulated by pleural mesothelial cells in tuberculous pleurisy. Am. J. Respir. Crit. Care Med. 2012, 185, 660–669.
- Ardain, A.; Domingo-Gonzalez, R.; Das, S.; Kazer, S.W.; Howard, N.C.; Singh, A.; Ahmed, M.; Nhamoyebonde, S.; Rangel-Moreno, J.; Ogongo, P.; et al. Group 3 innate lymphoid cells mediate early protective immunity against tuberculosis. Nature 2019, 570, 528–532.
- Dhiman, R.; Venkatasubramanian, S.; Paidipally, P.; Barnes, P.F.; Tvinnereim, A.; Vankayalapati, R. Interleukin 22 inhibits intracellular growth of Mycobacterium tuberculosis by enhancing calgranulin A expression. J. Infect. Dis. 2014, 209, 578–587.
- Dhiman, R.; Indramohan, M.; Barnes, P.F.; Nayak, R.C.; Paidipally, P.; Rao, L.V.; Vankayalapati, R. IL-22 produced by human NK cells inhibits growth of Mycobacterium tuberculosis by enhancing phagolysosomal fusion. J. Immunol. 2009, 183, 6639–6645.
- Hrabec, E.; Strek, M.; Zieba, M.; Kwiatkowska, S.; Hrabec, Z. Circulation level of matrix metalloproteinase-9 is correlated with disease severity in tuberculosis patients. Int. J. Tuberc. Lung Dis. Off. J. Int. Union Against Tuberc. Lung Dis. 2002, 6, 713–719.
- Backert, I.; Koralov, S.B.; Wirtz, S.; Kitowski, V.; Billmeier, U.; Martini, E.; Hofmann, K.; Hildner, K.; Wittkopf, N.; Brecht, K.; et al. STAT3 activation in Th17 and Th22 cells controls IL-22-mediated epithelial host defense during infectious colitis. J. Immunol. 2014, 193, 3779–3791.
- Basu, R.; O’Quinn, D.B.; Silberger, D.J.; Schoeb, T.R.; Fouser, L.; Ouyang, W.; Hatton, R.D.; Weaver, C.T. Th22 cells are an important source of IL-22 for host protection against enteropathogenic bacteria. Immunity 2012, 37, 1061–1075.
- Brandl, K.; Plitas, G.; Mihu, C.N.; Ubeda, C.; Jia, T.; Fleisher, M.; Schnabl, B.; DeMatteo, R.P.; Pamer, E.G. Vancomycin-resistant enterococci exploit antibiotic-induced innate immune deficits. Nature 2008, 455, 804–807.
- Kinnebrew, M.A.; Buffie, C.G.; Diehl, G.E.; Zenewicz, L.A.; Leiner, I.; Hohl, T.M.; Flavell, R.A.; Littman, D.R.; Pamer, E.G. Interleukin 23 production by intestinal CD103(+)CD11b(+) dendritic cells in response to bacterial flagellin enhances mucosal innate immune defense. Immunity 2012, 36, 276–287.
- Kinnebrew, M.A.; Ubeda, C.; Zenewicz, L.A.; Smith, N.; Flavell, R.A.; Pamer, E.G. Bacterial flagellin stimulates Toll-like receptor 5-dependent defense against vancomycin-resistant Enterococcus infection. J. Infect. Dis. 2010, 201, 534–543.
- Van Maele, L.; Carnoy, C.; Cayet, D.; Ivanov, S.; Porte, R.; Deruy, E.; Chabalgoity, J.A.; Renauld, J.C.; Eberl, G.; Benecke, A.G.; et al. Activation of Type 3 innate lymphoid cells and interleukin 22 secretion in the lungs during Streptococcus pneumoniae infection. J. Infect. Dis. 2014, 210, 493–503.
- Trevejo-Nunez, G.; Elsegeiny, W.; Conboy, P.; Chen, K.; Kolls, J.K. Critical Role of IL-22/IL22-RA1 Signaling in Pneumococcal Pneumonia. J. Immunol. 2016, 197, 1877–1883.
- Dixon, B.; Hossain, R.; Patel, R.V.; Algood, H.M.S. Th17 Cells in Helicobacter pylori Infection: A Dichotomy of Help and Harm. Infect. Immun. 2019, 87, e00363-19.
- Suerbaum, S.; Michetti, P. Helicobacter pylori infection. N. Engl. J. Med. 2002, 347, 1175–1186.
- Zhuang, Y.; Cheng, P.; Liu, X.F.; Peng, L.S.; Li, B.S.; Wang, T.T.; Chen, N.; Li, W.H.; Shi, Y.; Chen, W.; et al. A pro-inflammatory role for Th22 cells in Helicobacter pylori-associated gastritis. Gut 2015, 64, 1368–1378.
- Lv, Y.P.; Cheng, P.; Zhang, J.Y.; Mao, F.Y.; Teng, Y.S.; Liu, Y.G.; Kong, H.; Wu, X.L.; Hao, C.J.; Han, B.; et al. Helicobacter pylori-induced matrix metallopeptidase-10 promotes gastric bacterial colonization and gastritis. Sci. Adv. 2019, 5, eaau6547.
- Dixon, B.R.; Radin, J.N.; Piazuelo, M.B.; Contreras, D.C.; Algood, H.M. IL-17a and IL-22 Induce Expression of Antimicrobials in Gastrointestinal Epithelial Cells and May Contribute to Epithelial Cell Defense against Helicobacter pylori. PLoS ONE 2016, 11, e0148514.
- Carroll, K.C.; Bartlett, J.G. Biology of Clostridium difficile: Implications for epidemiology and diagnosis. Annu. Rev. Microbiol. 2011, 65, 501–521.
- Hasegawa, M.; Yada, S.; Liu, M.Z.; Kamada, N.; Muñoz-Planillo, R.; Do, N.; Núñez, G.; Inohara, N. Interleukin-22 regulates the complement system to promote resistance against pathobionts after pathogen-induced intestinal damage. Immunity 2014, 41, 620–632.
- Nagao-Kitamoto, H.; Leslie, J.L.; Kitamoto, S.; Jin, C.; Thomsson, K.A.; Gillilland, M.G., 3rd; Kuffa, P.; Goto, Y.; Jenq, R.R.; Ishii, C.; et al. Interleukin-22-mediated host glycosylation prevents Clostridioides difficile infection by modulating the metabolic activity of the gut microbiota. Nat. Med. 2020, 26, 608–617.
- Gomez-Simmonds, A.; Greenman, M.; Sullivan, S.B.; Tanner, J.P.; Sowash, M.G.; Whittier, S.; Uhlemann, A.C. Population Structure of Klebsiella pneumoniae Causing Bloodstream Infections at a New York City Tertiary Care Hospital: Diversification of Multidrug-Resistant Isolates. J. Clin. Microbiol. 2015, 53, 2060–2067.
- Ahn, D.; Wickersham, M.; Riquelme, S.; Prince, A. The Effects of IFN-λ on Epithelial Barrier Function Contribute to Klebsiella pneumoniae ST258 Pneumonia. Am. J. Respir. Cell Mol. Biol. 2019, 60, 158–166.
- Flo, T.H.; Smith, K.D.; Sato, S.; Rodriguez, D.J.; Holmes, M.A.; Strong, R.K.; Akira, S.; Aderem, A. Lipocalin 2 mediates an innate immune response to bacterial infection by sequestrating iron. Nature 2004, 432, 917–921.
- Berger, T.; Togawa, A.; Duncan, G.S.; Elia, A.J.; You-Ten, A.; Wakeham, A.; Fong, H.E.; Cheung, C.C.; Mak, T.W. Lipocalin 2-deficient mice exhibit increased sensitivity to Escherichia coli infection but not to ischemia-reperfusion injury. Proc. Natl. Acad. Sci. USA 2006, 103, 1834–1839.
- Matthay, M.A.; Ware, L.B.; Zimmerman, G.A. The acute respiratory distress syndrome. J. Clin. Investig. 2012, 122, 2731–2740.
- Broquet, A.; Jacqueline, C.; Davieau, M.; Besbes, A.; Roquilly, A.; Martin, J.; Caillon, J.; Dumoutier, L.; Renauld, J.C.; Heslan, M.; et al. Interleukin-22 level is negatively correlated with neutrophil recruitment in the lungs in a Pseudomonas aeruginosa pneumonia model. Sci. Rep. 2017, 7, 11010.
- Broquet, A.; Besbes, A.; Martin, J.; Jacqueline, C.; Vourc’h, M.; Roquilly, A.; Caillon, J.; Josien, R.; Asehnoune, K. Interleukin-22 regulates interferon lambda expression in a mice model of pseudomonas aeruginosa pneumonia. Mol. Immunol. 2020, 118, 52–59.
- Guillon, A.; Brea, D.; Morello, E.; Tang, A.; Jouan, Y.; Ramphal, R.; Korkmaz, B.; Perez-Cruz, M.; Trottein, F.; O’Callaghan, R.J.; et al. Pseudomonas aeruginosa proteolytically alters the interleukin 22-dependent lung mucosal defense. Virulence 2017, 8, 810–820.
- Guillon, A.; Brea, D.; Luczka, E.; Hervé, V.; Hasanat, S.; Thorey, C.; Pérez-Cruz, M.; Hordeaux, J.; Mankikian, J.; Gosset, P.; et al. Inactivation of the interleukin-22 pathway in the airways of cystic fibrosis patients. Cytokine 2019, 113, 470–474.
- Hohmann, E.L. Nontyphoidal salmonellosis. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2001, 32, 263–269.
- Raffatellu, M.; George, M.D.; Akiyama, Y.; Hornsby, M.J.; Nuccio, S.P.; Paixao, T.A.; Butler, B.P.; Chu, H.; Santos, R.L.; Berger, T.; et al. Lipocalin-2 resistance confers an advantage to Salmonella enterica serotype Typhimurium for growth and survival in the inflamed intestine. Cell Host Microbe 2009, 5, 476–486.
- Liu, J.Z.; Jellbauer, S.; Poe, A.J.; Ton, V.; Pesciaroli, M.; Kehl-Fie, T.E.; Restrepo, N.A.; Hosking, M.P.; Edwards, R.A.; Battistoni, A.; et al. Zinc sequestration by the neutrophil protein calprotectin enhances Salmonella growth in the inflamed gut. Cell Host Microbe 2012, 11, 227–239.
- Kehl-Fie, T.E.; Skaar, E.P. Nutritional immunity beyond iron: A role for manganese and zinc. Curr. Opin. Chem. Biol. 2010, 14, 218–224.
- Xiong, L.; Wang, S.; Dean, J.W.; Oliff, K.N.; Jobin, C.; Curtiss, R., 3rd; Zhou, L. Group 3 innate lymphoid cell pyroptosis represents a host defence mechanism against Salmonella infection. Nat. Microbiol. 2022, 7, 1087–1099.