Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Conner Chen and Version 1 by Takayuki Morimoto.
Glioblastoma (GBM) is the most aggressive and malignant primary brain tumor in adults. Despite multimodality treatment involving surgical resection, radiation therapy, chemotherapy, and tumor-treating fields, the median overall survival (OS) after diagnosis is approximately 2 years and the 5-year OS is poor. Considering the poor prognosis, novel treatment strategies are needed, such as immunotherapies. Natural killer (NK) cell-based immunotherapy involves the new feature of recognizing GBM via differing mechanisms from that of T cell-based immunotherapy.
- glioblastoma
- NK cell
- immunotherapy
1. Introduction
Glioblastoma (GBM) is the most common and aggressive primary brain tumor. The annual incidence of GBM is 3.19 per 100,000 [1] and it is classified as grade IV by the World Health Organization [2]. For several decades, the standard GBM therapy has consisted of maximum safety resection, adjuvant radiotherapy, and chemotherapy with temozolomide, termed the Stupp regimen. Despite the multidisciplinary therapy, the median overall survival (mOS) is only 15–17 months and the 5-year overall relative survival is only 5.8% [3,4][3][4]. Several novel strategies were investigated, where the addition of tumor-treating fields to standard treatment statistically significantly improved progression-free survival and overall survival (OS) [5]. A recent phase 2 trial of intratumoral oncolytic herpes virus G47∆ for residual or recurrent GBM demonstrated survival benefits and a good safety profile, which led to the approval of G47∆, albeit with conditions under the early approval system of Japanese-specific law, as the first oncolytic virus product in Japan [6]. Figure 1 summarizes the multimodality treatment against GBM. However, given the poor prognosis of patients with GBM, further novel approaches are needed for GBM treatment.
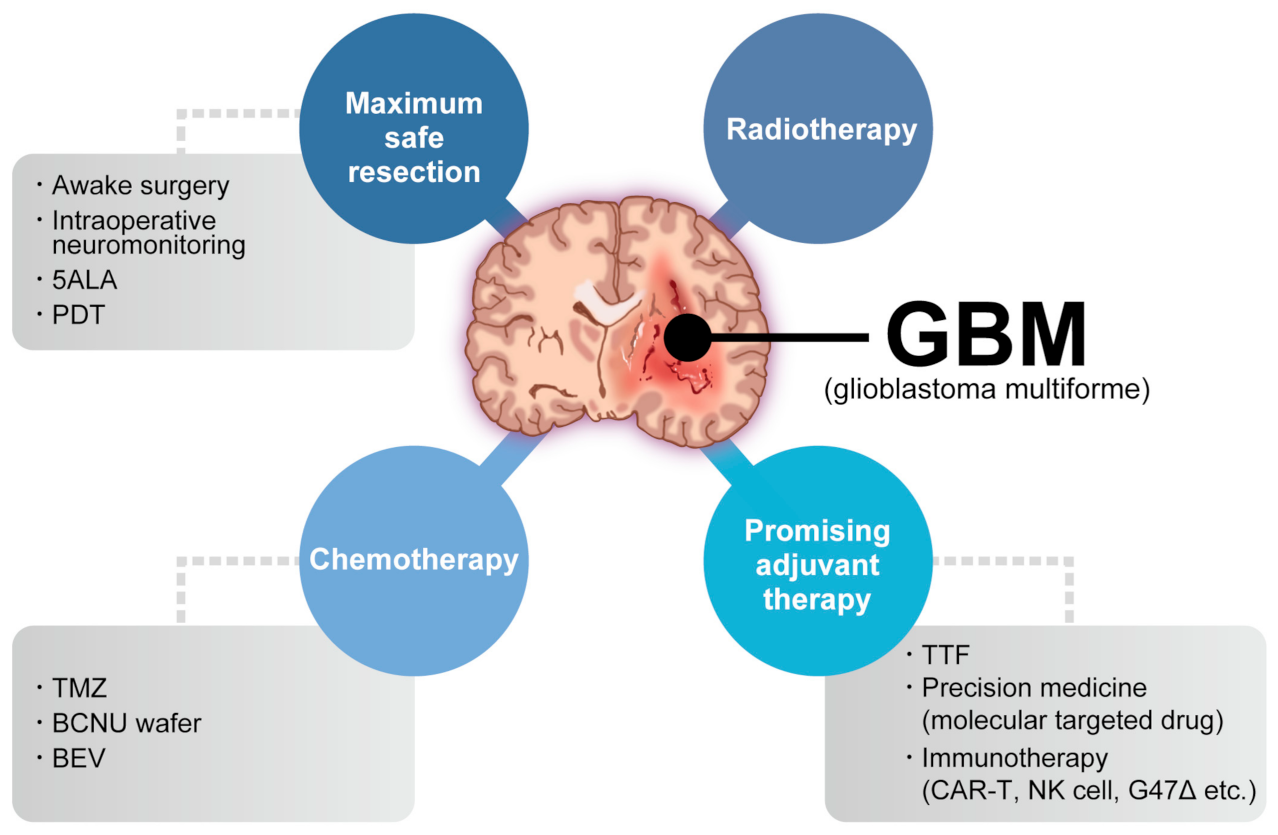
Figure 1. Multimodality treatment against GBM. While the standard therapy consists of maximum safety resection, adjuvant radiotherapy, and chemotherapy with temozolomide, which is collectively termed the Stupp regimen, several promising adjuvant therapies have been used for treating GBM. 5ALA: aminolevulinic acid, PDT: photodynamic therapy, TTF: tumor-treating fields, TMZ: temozolomide, BCNU: carmustine, BEV: bevacizumab.
Almost every immunotherapy mainly aims to generate a tumor-specific immune response to selectively eliminate tumor cells as a result of T cell activation. The representative immunotherapies are checkpoint inhibitor and chimeric antigen receptor (CAR) T-cell therapies, which have been used in other solid tumors and hematologic malignancies [7,8,9][7][8][9]. Contrastingly, several GBM immunotherapies have long been investigated, but few attractive strategies have been reported.
Compared to T cell-based therapy, natural killer (NK) cell-based therapy approaches tumors from new aspects. First, NK cells recognize the tumor, which consists of heterogeneous cells, via multiple activating and inhibitory receptors despite the diminished or absent expression of major histocompatibility complex class I (MHC-I) molecules [10]. Second, NK cells are important for recruiting conventional type 1 dendritic cells (cDC1s) and subsequently CD8+ T cells [11,12][11][12]. These functions promote cancer immunity cycle activation, which has the advantage of overcoming the immunosuppressive GBM tumor microenvironment (TME) [13,14][13][14].
2. NK Cell and NK Cell-Based Immunotherapy for Cancer
More than 40 years ago, it was determined that NK cells recognize cancer cells in mice and humans without antigen sensitization [58,59,60,61,62][15][16][17][18][19]. Recent research focused on the potential of NK cells in cell-based therapies. NK cells are considered an important part of the immune system, where they control microbial infections and tumor progression [58,63][15][20]. In patients and animal models, NK cell deficiency or impairment led to recurring virus infections and increased incidence of various types of cancer. In particular, NK cells controlled transplantable tumor growth and metastasis in numerous mouse models via antibody depletion of NK cells [64][21]. Additionally, NK cells are the founding members of the innate lymphoid cell (ILC) family [65][22]. In human peripheral blood, bone marrow, and tissues, NK cells can be identified by the expression of neural cell adhesion molecule (NCAM: CD56) and the absence of T cell receptor (TCR) and CD3 [66][23]. In the bone marrow, human NK cells derive from CD34+ hematopoietic progenitors and mature in the lymphoid organs [67,68][24][25]. Despite the differentiation from progenitor cells, NK cells persist in peripheral blood [69,70][26][27]. Human NK cell turnover in blood occurs over approximately 2 weeks [71][28]. NK cells can recognize tumor cells based on a balance between stimulatory and inhibitory receptors [stimulatory receptors: DNAX accessory molecule 1 (DNAM1), 2B4 (also known as CD244) and NK group 2D (NKG2D); inhibitory receptors: killer cell immunoglobulin-like receptors (KIRs), T-cell immunoreceptor with immunoglobulin and IGITTIM domains (TIGIT), killer cell lectin-like receptor subfamily G member 1 (KLRG1), T-cell immunoglobulin mucin family member 3 (TIM3), and programmed death 1 (PD1)] [72,73][29][30] (Figure 2). In detail, the main activating receptors are natural cytotoxicity receptors (NCRs: NKp-46/NCR1, NKp44/NCR2, NKp30/NCR3) [74,75,76,77][31][32][33][34]. B7-H6 and BAG6/BAT3 represent NKp30 ligands [78,79][35][36]. NKp44 recognizes a specific human leukocyte antigen (HLA)-DP molecule (HLA-DP401) and PCNA [80,81][37][38]. Barrow et al. also reported that platelet-derived growth factor (PDGF)-DD engagement of NKp44 triggered NK cell secretion of interferon (IFN)-γ and tumor necrosis factor alpha (TNF-α), and a distinctive transcriptional signature of PDGF-DD-induced cytokines and the downregulation of tumor cell-cycle genes correlated with NCR2 expression and greater survival in glioblastoma [82][39]. Gaggero et al. identified the extracellular matrix protein nidogen-1 (NID1) as a ligand of NKp44 [83][40]. NKp46 binds to the soluble plasma glycoprotein complement factor P/properdin [84][41]. Garg et al. reported vimentin (a 57-kDA molecule) as a putative NKp46 ligand [85][42]. The lysis of influenza virus (IV)-infected cells is mediated by the interaction between NKp46, and the IV hemagglutinin (HA) type 1 expressed by the infected cells [86,87,88][43][44][45]. NKG2D is another important NK receptor that transduces activating signals from the transmembrane adaptor protein DAP10 and recognizes UL16-binding proteins (ULBPs) and MHC class-1 related chain (MIC) A/B [89][46].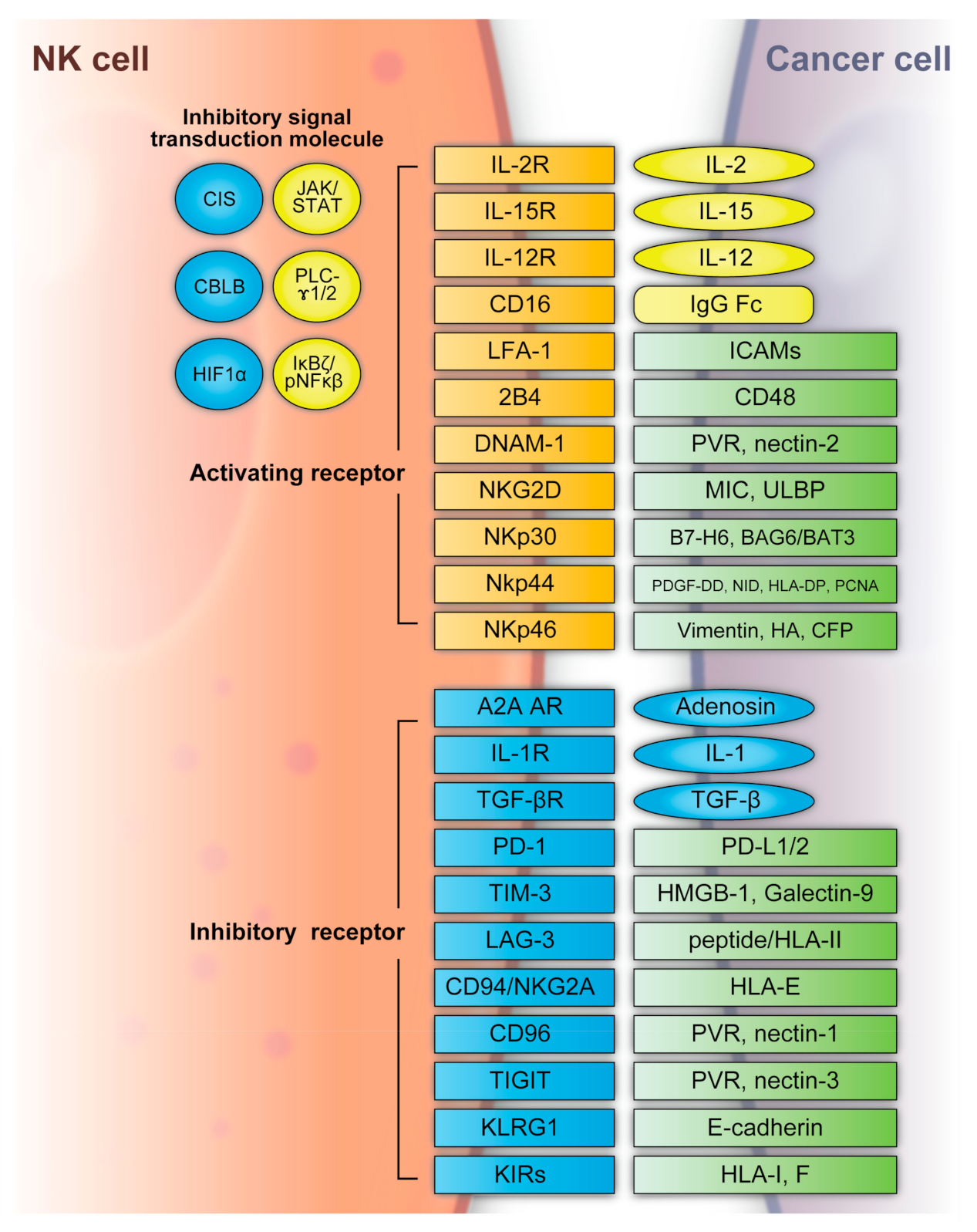
Figure 2. NK cell activating and inhibitory receptors. NK cells recognize tumor cells based on a balance between the stimulatory and inhibitory receptors above. A2A AR: A2A adenosine receptor, BAG6/BAT3: BCL2-associated athanogene cochaperone 6, CBLB: Casitas B-lineage lymphoma pro-oncogene-b, CIS: cytokine-inducible SH2-containing protein, DNAM1: DNAX accessory molecule 1, HA: IV hemagglutinin, HIF1α: hypoxia-inducible factor 1α, HLA: human leukocyte antigen, HLA-DP: human leukocyte antigen DP molecule, HMGB1: high-mobility group protein 1, ICAMs: intracellular adhesion molecules, IgG Fc: constant region of immunoglobulin, IL: interleukin, KIR: killer cell immunoglobulin-like receptors, JAK/STAT: Janus kinase/signal transducer and activator of transcription, KLRG: killer cell lectin-like receptor subfamily G member 1, LAG3: lymphocyte-activation gene 3, LFA-1: lymphocyte function-associated antigen-1, MIC: major histocompatibility complex class 1-related chain, NF-κB: nuclear factor kappa B, NID: nidogen, NKG2D: NK group 2D, NKp30: natural killer cell p30-related protein, NKp44: natural killer cell p44-related protein, NKp46: natural killer cell p46-related protein, PCNA: proliferating cell nuclear antigen, PD-1: programmed cell death 1, PDGF-DD: platelet-derived growth factor, PD-L1/2: programmed cell death ligand 1/2, PLC-γ1/2: phospholipase C γ1/2, PVR: poliovirus receptor, TGFβ: transforming growth factor β, TIGIT: T-cell immunoreceptor with immunoglobulin and ITIM domains, TIM3: T-cell immunoglobulin mucin family member 3, ULBP: UL16-binding protein.
References
- Ostrom, Q.T.; Gittleman, H.; Xu, J.; Kromer, C.; Wolinsky, Y.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2009–2013. Neuro. Oncol. 2016, 18, v1–v75.
- Osborn, A.G.; Louis, D.N.; Poussaint, T.Y.; Linscott, L.L.; Salzman, K.L. The 2021 World Health Organization Classification of Tumors of the Central Nervous System: What Neuroradiologists Need to Know. Am. J. Neuroradiol. 2022, 43, 928–937.
- Stupp, R.; Mason, W.P.; Van Den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996.
- Ostrom, Q.T.; Cote, D.J.; Ascha, M.; Kruchko, C.; Barnholtz-Sloan, J.S. Adult Glioma Incidence and Survival by Race or Ethnicity in the United States From 2000 to 2014. JAMA Oncol. 2018, 4, 1254.
- Stupp, R.; Taillibert, S.; Kanner, A.; Read, W.; Steinberg, D.M.; Lhermitte, B.; Toms, S.; Idbaih, A.; Ahluwalia, M.S.; Fink, K.; et al. Effect of Tumor-Treating Fields Plus Maintenance Temozolomide vs Maintenance Temozolomide Alone on Survival in Patients with Glioblastoma. JAMA 2017, 318, 2306.
- Todo, T.; Ito, H.; Ino, Y.; Ohtsu, H.; Ota, Y.; Shibahara, J.; Tanaka, M. Intratumoral oncolytic herpes virus G47∆ for residual or recurrent glioblastoma: A phase 2 trial. Nat. Med. 2022, 28, 1630–1639.
- Gandhi, L.; Rodríguez-Abreu, D.; Gadgeel, S.; Esteban, E.; Felip, E.; De Angelis, F.; Domine, M.; Clingan, P.; Hochmair, M.J.; Powell, S.F.; et al. Pembrolizumab plus Chemotherapy in Metastatic Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 2078–2092.
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.-J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2019, 381, 1535–1546.
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544.
- Vivier, E.; Tomasello, E.; Baratin, M.; Wsalzer, T.; Ugolini, S. Functions of natural killer cells. Nat. Immunol. 2008, 9, 503–510.
- Böttcher, J.P.; Bonavita, E.; Chakravarty, P.; Blees, H.; Cabeza-Cabrerizo, M.; Sammicheli, S.; Rogers, N.C.; Sahai, E.; Zelenay, S.; Reis E Sousa, C. NK Cells Stimulate Recruitment of cDC1 into the Tumor Microenvironment Promoting Cancer Immune Control. Cell 2018, 172, 1022–1037.e1014.
- Zhou, X.; Yu, J.; Cheng, X.; Zhao, B.; Manyam, G.C.; Zhang, L.; Schluns, K.; Li, P.; Wang, J.; Sun, S.-C. The deubiquitinase Otub1 controls the activation of CD8+ T cells and NK cells by regulating IL-15-mediated priming. Nat. Immunol. 2019, 20, 879–889.
- Klemm, F.; Maas, R.R.; Bowman, R.L.; Kornete, M.; Soukup, K.; Nassiri, S.; Brouland, J.-P.; Iacobuzio-Donahue, C.A.; Brennan, C.; Tabar, V.; et al. Interrogation of the Microenvironmental Landscape in Brain Tumors Reveals Disease-Specific Alterations of Immune Cells. Cell 2020, 181, 1643–1660.e1617.
- Grabowski, M.M.; Sankey, E.W.; Ryan, K.J.; Chongsathidkiet, P.; Lorrey, S.J.; Wilkinson, D.S.; Fecci, P.E. Immune suppression in gliomas. J. Neuro.-Oncol. 2021, 151, 3–12.
- Herberman, R.B.; Nunn, M.E.; Lavrin, D.H. Natural cytotoxic reactivity of mouse lymphoid cells against syngeneic and allogeneic tumors. I. Distribution of reactivity and specificity. Int. J. Cancer 1975, 16, 216–229.
- Roder, J.C.; Kiessling, R.; Biberfeld, P.; Andersson, B. Target-effector interaction in the natural killer (NK) cell system. II. The isolation of NK cells and studies on the mechanism of killing. J. Immunol. 1978, 121, 2509–2517.
- Hersey, P.; Edwards, A.; Edwards, J.; Adams, E.; Milton, G.W.; Nelson, D.S. Specificity of cell-mediated cytotoxicity against human melanoma lines: Evidence for “non-specific” killing by activated T-cells. Int. J. Cancer 1975, 16, 173–183.
- Peter, H.H.; Pavie-Fischer, J.; Fridman, W.H.; Aubert, C.; Cesarini, J.P.; Roubin, R.; Kourilsky, F.M. Cell-mediate cytotoxicity in vitro of human lymphocytes against a tissue culture melanoma cell line (igr3). J. Immunol. 1975, 115, 539–548.
- West, W.H.; Cannon, G.B.; Kay, H.D.; Bonnard, G.D.; Herberman, R.B. Natural cytotoxic reactivity of human lymphocytes against a myeloid cell line: Characterization of effector cells. J. Immunol. 1977, 118, 355–361.
- Kiessling, R.; Klein, E.; Wigzell, H. “Natural” killer cells in the mouse. I. Cytotoxic cells with specificity for mouse Moloney leukemia cells. Specificity and distribution according to genotype. Eur. J. Immunol. 1975, 5, 112–117.
- Smyth, M.J.; Hayakawa, Y.; Takeda, K.; Yagita, H. New aspects of natural-killer-cell surveillance and therapy of cancer. Nat. Rev. Cancer 2002, 2, 850–861.
- Artis, D.; Spits, H. The biology of innate lymphoid cells. Nature 2015, 517, 293–301.
- Lanier, L.L.; Testi, R.; Bindl, J.; Phillips, J.H. Identity of Leu-19 (CD56) leukocyte differentiation antigen and neural cell adhesion molecule. J. Exp. Med. 1989, 169, 2233–2238.
- Yu, J.; Freud, A.G.; Caligiuri, M.A. Location and cellular stages of natural killer cell development. Trends Immunol. 2013, 34, 573–582.
- Virginie; Zriwil, A.; Peitzsch, C.; Michaëlsson, J.; Friberg, D.; Soneji, S.; Sitnicka, E. Identification of a Human Natural Killer Cell Lineage-Restricted Progenitor in Fetal and Adult Tissues. Immunity 2015, 43, 394–407.
- Schlums, H.; Jung, M.; Han, H.; Theorell, J.; Bigley, V.; Chiang, S.C.; Allan, D.S.; Davidson-Moncada, J.K.; Dickinson, R.E.; Holmes, T.D.; et al. Adaptive NK cells can persist in patients with GATA2 mutation depleted of stem and progenitor cells. Blood 2017, 129, 1927–1939.
- Corat, M.A.F.; Schlums, H.; Wu, C.; Theorell, J.; Espinoza, D.A.; Sellers, S.E.; Townsley, D.M.; Young, N.S.; Bryceson, Y.T.; Dunbar, C.E.; et al. Acquired somatic mutations in PNH reveal long-term maintenance of adaptive NK cells independent of HSPCs. Blood 2017, 129, 1940–1946.
- Zhang, Y.; Wallace, D.L.; De Lara, C.M.; Ghattas, H.; Asquith, B.; Worth, A.; Griffin, G.E.; Taylor, G.P.; Tough, D.F.; Beverley, P.C.L.; et al. In vivo kinetics of human natural killer cells: The effects of ageing and acute and chronic viral infection. Immunology 2007, 121, 258–265.
- Morvan, M.G.; Lanier, L.L. NK cells and cancer: You can teach innate cells new tricks. Nat. Rev. Cancer 2016, 16, 7–19.
- Guillerey, C.; Huntington, N.D.; Smyth, M.J. Targeting natural killer cells in cancer immunotherapy. Nat. Immunol. 2016, 17, 1025–1036.
- Moretta, A.; Bottino, C.; Vitale, M.; Pende, D.; Cantoni, C.; Mingari, M.C.; Biassoni, R.; Moretta, L. Activating receptors and coreceptors involved in human natural killer cell-mediated cytolysis. Annu. Rev. Immunol. 2001, 19, 197–223.
- Pessino, A.; Sivori, S.; Bottino, C.; Malaspina, A.; Morelli, L.; Moretta, L.; Biassoni, R.; Moretta, A. Molecular cloning of NKp46: A novel member of the immunoglobulin superfamily involved in triggering of natural cytotoxicity. J. Exp. Med. 1998, 188, 953–960.
- Vitale, M.; Bottino, C.; Sivori, S.; Sanseverino, L.; Castriconi, R.; Marcenaro, E.; Augugliaro, R.; Moretta, L.; Moretta, A. NKp44, a novel triggering surface molecule specifically expressed by activated natural killer cells, is involved in non-major histocompatibility complex-restricted tumor cell lysis. J. Exp. Med. 1998, 187, 2065–2072.
- Pende, D.; Parolini, S.; Pessino, A.; Sivori, S.; Augugliaro, R.; Morelli, L.; Marcenaro, E.; Accame, L.; Malaspina, A.; Biassoni, R.; et al. Identification and molecular characterization of NKp30, a novel triggering receptor involved in natural cytotoxicity mediated by human natural killer cells. J. Exp. Med. 1999, 190, 1505–1516.
- Brandt, C.S.; Baratin, M.; Yi, E.C.; Kennedy, J.; Gao, Z.; Fox, B.; Haldeman, B.; Ostrander, C.D.; Kaifu, T.; Chabannon, C.; et al. The B7 family member B7-H6 is a tumor cell ligand for the activating natural killer cell receptor NKp30 in humans. J. Exp. Med. 2009, 206, 1495–1503.
- Binici, J.; Hartmann, J.; Herrmann, J.; Schreiber, C.; Beyer, S.; Güler, G.; Vogel, V.; Tumulka, F.; Abele, R.; Mäntele, W.; et al. A Soluble Fragment of the Tumor Antigen BCL2-associated Athanogene 6 (BAG-6) Is Essential and Sufficient for Inhibition of NKp30 Receptor-dependent Cytotoxicity of Natural Killer Cells. J. Biol. Chem. 2013, 288, 34295–34303.
- Rosental, B.; Brusilovsky, M.; Hadad, U.; Oz, D.; Appel, M.Y.; Afergan, F.; Yossef, R.; Rosenberg, L.A.; Aharoni, A.; Cerwenka, A.; et al. Proliferating Cell Nuclear Antigen Is a Novel Inhibitory Ligand for the Natural Cytotoxicity Receptor NKp44. J. Immunol. 2011, 187, 5693–5702.
- Niehrs, A.; Garcia-Beltran, W.F.; Norman, P.J.; Watson, G.M.; Hölzemer, A.; Chapel, A.; Richert, L.; Pommerening-Röser, A.; Körner, C.; Ozawa, M.; et al. A subset of HLA-DP molecules serve as ligands for the natural cytotoxicity receptor NKp44. Nat. Immunol. 2019, 20, 1129–1137.
- Barrow, A.D.; Edeling, M.A.; Trifonov, V.; Luo, J.; Goyal, P.; Bohl, B.; Bando, J.K.; Kim, A.H.; Walker, J.; Andahazy, M.; et al. Natural Killer Cells Control Tumor Growth by Sensing a Growth Factor. Cell 2018, 172, 534–548.e519.
- Gaggero, S.; Bruschi, M.; Petretto, A.; Parodi, M.; Zotto, G.D.; Lavarello, C.; Prato, C.; Santucci, L.; Barbuto, A.; Bottino, C.; et al. Nidogen-1 is a novel extracellular ligand for the NKp44 activating receptor. OncoImmunology 2018, 7, e1470730.
- Narni-Mancinelli, E.; Gauthier, L.; Baratin, M.; Guia, S.; Fenis, A.; Deghmane, A.-E.; Rossi, B.; Fourquet, P.; Escalière, B.; Kerdiles, Y.M.; et al. Complement factor P is a ligand for the natural killer cell–activating receptor NKp46. Sci. Immunol. 2017, 2, eaam9628.
- Garg, A.; Barnes, P.F.; Porgador, A.; Roy, S.; Wu, S.; Nanda, J.S.; Griffith, D.E.; Girard, W.M.; Rawal, N.; Shetty, S.; et al. Vimentin Expressed on Mycobacterium tuberculosis-Infected Human Monocytes Is Involved in Binding to the NKp46 Receptor1. J. Immunol. 2006, 177, 6192–6198.
- Mendelson, M.; Tekoah, Y.; Zilka, A.; Gershoni-Yahalom, O.; Gazit, R.; Achdout, H.; Bovin, N.V.; Meningher, T.; Mandelboim, M.; Mandelboim, O.; et al. NKp46 O-Glycan Sequences That Are Involved in the Interaction with Hemagglutinin Type 1 of Influenza Virus. J. Virol. 2010, 84, 3789–3797.
- Arnon, T.I.; Achdout, H.; Lieberman, N.; Gazit, R.; Gonen-Gross, T.; Katz, G.; Bar-Ilan, A.; Bloushtain, N.; Lev, M.; Joseph, A.; et al. The mechanisms controlling the recognition of tumor- and virus-infected cells by NKp46. Blood 2004, 103, 664–672.
- Mandelboim, O.; Lieberman, N.; Lev, M.; Paul, L.; Arnon, T.I.; Bushkin, Y.; Davis, D.M.; Strominger, J.L.; Yewdell, J.W.; Porgador, A. Recognition of haemagglutinins on virus-infected cells by NKp46 activates lysis by human NK cells. Nature 2001, 409, 1055–1060.
- Bauer, S.; Groh, V.; Wu, J.; Steinle, A.; Phillips, J.H.; Lanier, L.L.; Spies, T. Activation of NK Cells and T Cells by NKG2D, a Receptor for Stress-Inducible MICA. Science 1999, 285, 727–729.
- López-Botet, M.; Llano, M.; Navarro, F.; Bellón, T. NK cell recognition of non-classical HLA class I molecules. Semin. Immunol. 2000, 12, 109–119.
- Lanier, L.L.; Corliss, B.; Wu, J.; Phillips, J.H. Association of DAP12 with Activating CD94/NKG2C NK Cell Receptors. Immunity 1998, 8, 693–701.
- Sivori, S.; Vacca, P.; Del Zotto, G.; Munari, E.; Mingari, M.C.; Moretta, L. Human NK cells: Surface receptors, inhibitory checkpoints, and translational applications. Cell. Mol. Immunol. 2019, 16, 430–441.
- Huntington, N.D.; Tabarias, H.; Fairfax, K.; Brady, J.; Hayakawa, Y.; Degli-Esposti, M.A.; Smyth, M.J.; Tarlinton, D.M.; Nutt, S.L. NK Cell Maturation and Peripheral Homeostasis Is Associated with KLRG1 Up-Regulation1. J. Immunol. 2007, 178, 4764–4770.
- Sivori, S.; Parolini, S.; Falco, M.; Marcenaro, E.; Biassoni, R.; Bottino, C.; Moretta, L.; Moretta, A. 2B4 functions as a co-receptor in human NK cell activation. Eur. J. Immunol. 2000, 30, 787–793.
- Shibuya, A.; Campbell, D.; Hannum, C.; Yssel, H.; Franz-Bacon, K.; McClanahan, T.; Kitamura, T.; Nicholl, J.; Sutherland, G.R.; Lanier, L.L.; et al. DNAM-1, A Novel Adhesion Molecule Involved in the Cytolytic Function of T Lymphocytes. Immunity 1996, 4, 573–581.
- Kärre, K.; Ljunggren, H.G.; Piontek, G.; Kiessling, R. Selective rejection of H–2-deficient lymphoma variants suggests alternative immune defence strategy. Nature 1986, 319, 675–678.
- Vivier, E.; Ugolini, S.; Blaise, D.; Chabannon, C.; Brossay, L. Targeting natural killer cells and natural killer T cells in cancer. Nat. Rev. Immunol. 2012, 12, 239–252.
- Freud, A.G.; Mundy-Bosse, B.L.; Yu, J.; Caligiuri, M.A. The Broad Spectrum of Human Natural Killer Cell Diversity. Immunity 2017, 47, 820–833.
- Raulet, D.H.; Vance, R.E. Self-tolerance of natural killer cells. Nat. Rev. Immunol. 2006, 6, 520–531.
- Elliott, J.M.; Yokoyama, W.M. Unifying concepts of MHC-dependent natural killer cell education. Trends Immunol. 2011, 32, 364–372.
- Goodridge, J.P.; Burian, A.; Lee, N.; Geraghty, D.E. HLA-F and MHC class I open conformers are ligands for NK cell Ig-like receptors. J. Immunol. 2013, 191, 3553–3562.
- Ferris, R.L.; Jaffee, E.M.; Ferrone, S. Tumor Antigen–Targeted, Monoclonal Antibody–Based Immunotherapy: Clinical Response, Cellular Immunity, and Immunoescape. J. Clin. Oncol. 2010, 28, 4390–4399.
- Bournazos, S.; Wang, T.T.; Dahan, R.; Maamary, J.; Ravetch, J.V. Signaling by Antibodies: Recent Progress. Annu. Rev. Immunol. 2017, 35, 285–311.
- Delconte, R.B.; Kolesnik, T.B.; Dagley, L.F.; Rautela, J.; Shi, W.; Putz, E.M.; Stannard, K.; Zhang, J.-G.; Teh, C.; Firth, M.; et al. CIS is a potent checkpoint in NK cell–mediated tumor immunity. Nat. Immunol. 2016, 17, 816–824.
- Delconte, R.B.; Guittard, G.; Goh, W.; Hediyeh-Zadeh, S.; Hennessy, R.J.; Rautela, J.; Davis, M.J.; Souza-Fonseca-Guimaraes, F.; Nunès, J.A.; Huntington, N.D. NK Cell Priming from Endogenous Homeostatic Signals Is Modulated by CIS. Front. Immunol. 2020, 11, 75.
- Barsoum, I.B.; Hamilton, T.K.; Li, X.; Cotechini, T.; Miles, E.A.; Siemens, D.R.; Graham, C.H. Hypoxia induces escape from innate immunity in cancer cells via increased expression of ADAM10: Role of nitric oxide. Cancer Res. 2011, 71, 7433–7441.
- Balsamo, M.; Manzini, C.; Pietra, G.; Raggi, F.; Blengio, F.; Mingari, M.C.; Varesio, L.; Moretta, L.; Bosco, M.C.; Vitale, M. Hypoxia downregulates the expression of activating receptors involved in NK-cell-mediated target cell killing without affecting ADCC. Eur. J. Immunol. 2013, 43, 2756–2764.
- Krzywinska, E.; Kantari-Mimoun, C.; Kerdiles, Y.; Sobecki, M.; Isagawa, T.; Gotthardt, D.; Castells, M.; Haubold, J.; Millien, C.; Viel, T.; et al. Loss of HIF-1α in natural killer cells inhibits tumour growth by stimulating non-productive angiogenesis. Nat. Commun. 2017, 8, 1597.
- Ni, J.; Wang, X.; Stojanovic, A.; Zhang, Q.; Wincher, M.; Bühler, L.; Arnold, A.; Correia, M.P.; Winkler, M.; Koch, P.-S.; et al. Single-Cell RNA Sequencing of Tumor-Infiltrating NK Cells Reveals that Inhibition of Transcription Factor HIF-1α Unleashes NK Cell Activity. Immunity 2020, 52, 1075–1087.e1078.
- Lequeux, A.; Noman, M.Z.; Xiao, M.; Van Moer, K.; Hasmim, M.; Benoit, A.; Bosseler, M.; Viry, E.; Arakelian, T.; Berchem, G.; et al. Targeting HIF-1 alpha transcriptional activity drives cytotoxic immune effector cells into melanoma and improves combination immunotherapy. Oncogene 2021, 40, 4725–4735.
- Mohapatra, B.; Ahmad, G.; Nadeau, S.; Zutshi, N.; An, W.; Scheffe, S.; Dong, L.; Feng, D.; Goetz, B.; Arya, P.; et al. Protein tyrosine kinase regulation by ubiquitination: Critical roles of Cbl-family ubiquitin ligases. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2013, 1833, 122–139.
- Paolino, M.; Choidas, A.; Wallner, S.; Pranjic, B.; Uribesalgo, I.; Loeser, S.; Jamieson, A.M.; Langdon, W.Y.; Ikeda, F.; Fededa, J.P.; et al. The E3 ligase Cbl-b and TAM receptors regulate cancer metastasis via natural killer cells. Nature 2014, 507, 508–512.
- Matalon, O.; Barda-Saad, M. Cbl ubiquitin ligases mediate the inhibition of natural killer cell activity. Commun. Integr. Biol. 2016, 9, e1216739.
- Matalon, O.; Fried, S.; Ben-Shmuel, A.; Pauker, M.H.; Joseph, N.; Keizer, D.; Piterburg, M.; Barda-Saad, M. Dephosphorylation of the adaptor LAT and phospholipase C-γ by SHP-1 inhibits natural killer cell cytotoxicity. Sci. Signal. 2016, 9, ra54.
- Cerwenka, A.; Lanier, L.L. Natural killer cell memory in infection, inflammation and cancer. Nat. Rev. Immunol. 2016, 16, 112–123.
- Romee, R.; Schneider, S.E.; Leong, J.W.; Chase, J.M.; Keppel, C.R.; Sullivan, R.P.; Cooper, M.A.; Fehniger, T.A. Cytokine activation induces human memory-like NK cells. Blood 2012, 120, 4751–4760.
- Rückert, T.; Lareau, C.A.; Mashreghi, M.-F.; Ludwig, L.S.; Romagnani, C. Clonal expansion and epigenetic inheritance of long-lasting NK cell memory. Nat. Immunol. 2022, 23, 1551–1563.
- Orange, J.S. Formation and function of the lytic NK-cell immunological synapse. Nat. Rev. Immunol. 2008, 8, 713–725.
- Gwalani, L.A.; Orange, J.S. Single Degranulations in NK Cells Can Mediate Target Cell Killing. J. Immunol. 2018, 200, 3231–3243.
- Liu, D.; Bryceson, Y.T.; Meckel, T.; Vasiliver-Shamis, G.; Dustin, M.L.; Long, E.O. Integrin-Dependent Organization and Bidirectional Vesicular Traffic at Cytotoxic Immune Synapses. Immunity 2009, 31, 99–109.
- Rossin, A.; Miloro, G.; Hueber, A.-O. TRAIL and FasL Functions in Cancer and Autoimmune Diseases: Towards an Increasing Complexity. Cancers 2019, 11, 639.
- Betts, M.R.; Brenchley, J.M.; Price, D.A.; De Rosa, S.C.; Douek, D.C.; Roederer, M.; Koup, R.A. Sensitive and viable identification of antigen-specific CD8+ T cells by a flow cytometric assay for degranulation. J. Immunol. Methods 2003, 281, 65–78.
- Chiossone, L.; Dumas, P.Y.; Vienne, M.; Vivier, E. Natural killer cells and other innate lymphoid cells in cancer. Nat. Rev. Immunol. 2018, 18, 671–688.
- Barry, K.C.; Hsu, J.; Broz, M.L.; Cueto, F.J.; Binnewies, M.; Combes, A.J.; Nelson, A.E.; Loo, K.; Kumar, R.; Rosenblum, M.D.; et al. A natural killer–dendritic cell axis defines checkpoint therapy–responsive tumor microenvironments. Nat. Med. 2018, 24, 1178–1191.
- Cursons, J.; Souza-Fonseca-Guimaraes, F.; Foroutan, M.; Anderson, A.; Hollande, F.; Hediyeh-Zadeh, S.; Behren, A.; Huntington, N.D.; Davis, M.J. A Gene Signature Predicting Natural Killer Cell Infiltration and Improved Survival in Melanoma Patients. Cancer Immunol. Res. 2019, 7, 1162–1174.
- Schmidt, L.; Eskiocak, B.; Kohn, R.; Dang, C.; Joshi, N.S.; Dupage, M.; Lee, D.-Y.; Jacks, T. Enhanced adaptive immune responses in lung adenocarcinoma through natural killer cell stimulation. Proc. Natl. Acad. Sci. USA 2019, 116, 17460–17469.
- Zemek, R.M.; De Jong, E.; Chin, W.L.; Schuster, I.S.; Fear, V.S.; Casey, T.H.; Forbes, C.; Dart, S.J.; Leslie, C.; Zaitouny, A.; et al. Sensitization to immune checkpoint blockade through activation of a STAT1/NK axis in the tumor microenvironment. Sci. Transl. Med. 2019, 11, eaav7816.
- Spranger, S.; Dai, D.; Horton, B.; Gajewski, T.F. Tumor-Residing Batf3 Dendritic Cells Are Required for Effector T Cell Trafficking and Adoptive T Cell Therapy. Cancer Cell 2017, 31, 711–723.e714.
- Benci, J.L.; Johnson, L.R.; Choa, R.; Xu, Y.; Qiu, J.; Zhou, Z.; Xu, B.; Ye, D.; Nathanson, K.L.; June, C.H.; et al. Opposing Functions of Interferon Coordinate Adaptive and Innate Immune Responses to Cancer Immune Checkpoint Blockade. Cell 2019, 178, 933–948.e914.
- Ruggeri, L.; Capanni, M.; Urbani, E.; Perruccio, K.; Shlomchik, W.D.; Tosti, A.; Posati, S.; Rogaia, D.; Frassoni, F.; Aversa, F.; et al. Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science 2002, 295, 2097–2100.
- Miller, J.S.; Soignier, Y.; Panoskaltsis-Mortari, A.; McNearney, S.A.; Yun, G.H.; Fautsch, S.K.; McKenna, D.; Le, C.; Defor, T.E.; Burns, L.J.; et al. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood 2005, 105, 3051–3057.
- Cooley, S.; He, F.; Bachanova, V.; Vercellotti, G.M.; DeFor, T.E.; Curtsinger, J.M.; Robertson, P.; Grzywacz, B.; Conlon, K.C.; Waldmann, T.A.; et al. First-in-human trial of rhIL-15 and haploidentical natural killer cell therapy for advanced acute myeloid leukemia. Blood Adv. 2019, 3, 1970–1980.
- Romee, R.; Rosario, M.; Berrien-Elliott, M.M.; Wagner, J.A.; Jewell, B.A.; Schappe, T.; Leong, J.W.; Abdel-Latif, S.; Schneider, S.E.; Willey, S.; et al. Cytokine-induced memory-like natural killer cells exhibit enhanced responses against myeloid leukemia. Sci. Transl. Med. 2016, 8, 357ra123–357ra351.
- Berrien-Elliott, M.M.; Jacobs, M.T.; Fehniger, T.A. Allogeneic natural killer cell therapy. Blood 2022.
More