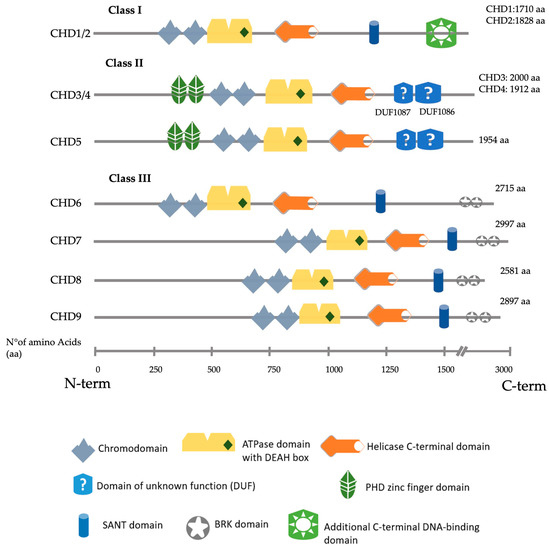
Figure 1. Schematic representation of all nine human CHD proteins divided by their class, showing the approximate position of the main predicted domains, as described in Uniprot, SMART and InterPro
[6][7][8][14,15,16].
2. CHD3/4
CHD3 and CDH4 share 71.6% of their amino acid sequences. Both proteins form distinct nucleosome remodelling deacetylase (NuRD) complexes with different but overlapping functionalities
[9][17]. Mass spectrometry data show that CHD3 and CHD4 may form heterodimers within NuRD complexes
[10][18]. CHD3 and CHD4 are the core subunit of the NuRD complex
[11][19]. Their binding to this complex is mutually exclusive
[12][20]. Normal NuRD complexes are essential for many developmental and cell differentiation processes, including maintaining embryonic stem (ES) cell pluripotency and regulating progenitor cell development in numerous organs such as the brain, hematopoietic system or kidney
[13][14][21,22].
Snijders Blok–Campeau syndrome is a recently described syndrome that encompasses patients with variants in
CHD3, characterised by intellectual disability, developmental delay (especially speech delay) and several dysmorphic features
[15][23], with most of the mutations localising within the ATPase or the helicase domain
[16][24]. A minority of these patients manifest neurosensory hearing loss (3 out of 24). More investigations are needed to understand further the implications of hearing deficit in this syndrome.
Mutations in the CHD4 protein result in Sifrim–Hitz–Weiss (SIHIWES) syndrome, an autosomal dominant neurodevelopmental disease. Symptoms identified include global developmental delay, mild to moderate intellectual disability, brain anomalies, congenital heart defects, dysmorphic features, macrocephaly and conductive and/or sensorineural hearing loss in 55% of the cases
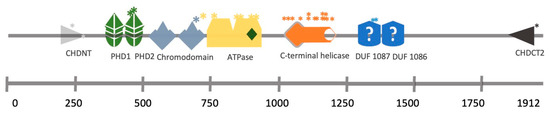
[17][25]. The mutations identified in hearing loss patients affect the ATPase domain, the PHD domain or the DUF1087 domain (
Figure 2)
[17][18][25,26].
Figure 2.
CHD4 structural domains and the location of SNPs identified in a cohort of SIHIWES patients.
*
Mutation position.
In mice, Chd4 is present in the organ of Corti, starting at embryonic day 18.5 (E18.5) throughout most cells. It remains present during postnatal development, and its expression is restricted to
supporting cells (SCs
) at the adult stage (postnatal P21)
[19][27]. In the central nervous system, Chd4 is strongly expressed in both neural progenitors and mature neurons. Conditional knockout of Chd4 in nestin-positive neural progenitors in the mouse cerebral cortex leads to their precocious cell cycle exit and apoptosis, thereby causing microcephaly
[20][28]. It has been suggested that Chd4 regulates neural differentiation by controlling the cell cycle through repressing the acetylation of p53 and possible transcription activation of neural differentiation genes
[14][22]. Whether Chd4 has the same effect in the cochlea remains to be established.
Altogether, these findings on
CHD4 mutations in hearing loss patients and the
Chd4 pattern of expression in embryonic mice cochlea suggest that CHD4 may influence spatiotemporal gene expression regulation in the developing cochlea. This deficiency could lead to deafness. Further experimental evaluation is critical to decode the mechanistic role of CHD4 in otic development and how particular missense mutations can give rise to the deafness phenotype.
3. CHD7
De novo heterozygous mutations in the
CHD7 gene are the leading causes of CHARGE syndrome, a complex neurodevelopmental disorder characterised by ocular
Coloboma,
Heart defects,
Atresia of the choanae,
Retarded growth and development,
Genital hypoplasia and
Ear abnormalities, including mixed conductive/sensorineural hearing loss
[21][22][29,30]. More than 500 heterozygous
CHD7 mutations have been identified in CHARGE syndrome patients, most of which are nonsense, frameshift indels, splice site and missense mutations
[23][31].
In mice, Chd7 is highly expressed throughout the developing otocyst starting at E9.5. Later during development, Chd7 is present in the cochlear epithelium both in
hair cells (HCs
) and SCs and in spiral ganglion neurons. Chd7 is still present postnatally but is decreased in the organ of Corti
[24][25][32,33].
A Chd7 heterozygous mouse model was generated from Chd7-deficient, gene-trapped lacZ reporter ES cells (hereafter Chd7
Gt/+ mice) that expresses β-galactosidase under the control of the Chd7 promoter
[26][34]. Homozygous Chd7
Gt/Gt are embryonic lethal. HC formation and cochlear innervation were normal in Chd7
Gt/+ mice. However, Chd7
Gt/+ animals present mild hearing loss with elevated auditory brainstem recordings (ABR) and reduced distortion product otoacoustic emissions (DPOAE)
[26][34]. Recently, a re-evaluation of Chd7
Gt/+ suggests a new function in the development and maintenance of satellite glial cells in the spiral ganglia, as well as in the regulation of myelin sheaths surrounding type I spiral ganglion neurons that innervate the sensory epithelium of the cochlea
[27][35]. In two ENU-induced mutations in Chd7—looper and trooper strains—having a nonsense mutation (c.5690C>A, p.S1897X) or a cryptic splice site mutation (c.3219-18T>A), respectively, middle ear and vestibular defects were found to be a prominent phenotype with early onset hearing loss
[28][29][36,37].
Deficiency in Chd7 during development has also been reported to specifically impact neuronal development in the inner ear. In a Foxg1-Cre-driven conditional Chd7 knockout mouse, the otocyst had a reduced vestibulocochlear ganglion size. This smaller neuronal number is due to the reduced expression of the neuronal fate-specific genes Ngn1, Otx2 and Fgf10, leading to reduced neuronal cell proliferation
[24][30][32,38]. A recent study has shown that Chd7 deficiency leads to IHC and auditory neurons being vulnerable to ototoxic stress, rapid postnatal degeneration and profound hearing loss
[31][39].
Modulating retinoic acid signalling prevents inner ear defects caused by Chd7 deficiency
[32][40]. Indeed, loss of the retinoic acid (RA) synthetic enzyme Aldh1a3 or treatment with citral—an inhibitor of
retinoic acid (RA
) synthesis—partially rescued the semicircular canal deformation phenotype in Chd7
Gt/+ mice
[33][41]. Upregulation or downregulation of RA signalling during embryogenesis leads to developmental defects similar to those observed in CHARGE. RNA-seq and qRT-PCR combined with ChIP experiments in inner ears demonstrate that Chd7 acts upstream of
Aldh1a3 via direct binding and repression of
Aldh1a3 [33][41].
To evaluate the Chd7 deficiency at single-cell resolution, Durruthy-Durruthy et al. performed single-cell multiplexed qPCR in Pax2Cre; mT/mGFP otocysts cells from Chd7+/+ and Chd7
Gt/+ mice for 192 inner ear specific genes and reported a cellular identity shift towards neuroblasts in E10.5 otocysts
[34][42]. This cell fate decision may arise through disruption in the pro-sensory and pro-neurogenic gene expression network and Notch signalling pathways.
To model the inner ear phenotypes in CHARGE syndrome in a human context, Nie et al. adopted a previously published human induced pluripotent stem (hiPS) cell/human ES cell-derived otic organoid model
[35][43]. The study reported a complete loss of inner ear HC formation in the CHD7 knock-out line (
CHD7−/−) or CHD7 patient-specific missense mutation (CHD7
S834F/S834F) d70 otic organoids
[36][44]. In contrast, the haploinsufficient/heterozygous otic organoids (CHD7
S834F/+ or CHD7
+/−) could generate HCs. To understand the molecular pathogenesis, the CHD7 mutations were created in a WA25 PAX2nGFP hESC line, which allows for the study of the transcriptome of
PAX2+ otic progenitors from both CHD7
+/+ and CHD7
−/− d20 organoids. Single-cell RNA-sequencing (scRNA-seq) transcriptome data on those PAX2+ cells revealed a downregulation of several essential inner ear morphogenesis and developmental genes such as
TBX1,
LMX1A,
SOX10,
DLX5 and
SIX1 and disruption in FGF, BMP, Notch, TGF-β and Wnt signalling pathways.
These effects on PAX2 otic progenitors were reflected in HC development. Indeed, scRNA-seq on POU4F3-nd tomato hair cells from
CHD7+/− d70 organoids showed numerous dysregulated deafness genes compared with the WT counterparts.
SIX1,
USH1C and
STRC were downregulated, giving potential explanations for hearing loss observed in patients with CHARGE syndrome. Co-differentiating both WT and
CHD7−/− hESCs in a chimeric organoid system partially restored the expression of otherwise severely downregulated essential otic genes
FBXO2,
SOX10 and
DLX5 but failed to generate any hair cells in the CHD7
−/− background
[36][44]. Altogether, this study described the critical role of CHD7 in otic lineage differentiation and hair cell development and, more importantly, in an
in-vitro humanised experimental model.
4. CHD8
CHD8 is most prominently known for its association with autism spectrum disorders (ASD)
[37][45]. CHD8 has two isoforms: CHD8L, a full-length version of the protein weighing 280 kDa, and CHD8S (Duplin), a 110 kDa protein of the NH2-terminal chromodomain region. However, the latter does not have a counterpart in humans
[38][46]. CHD8 interacts with CHD7 and FAM124B (family with sequence similarity 124B) as a complex
[39][40][47,48]. However, the functional implication of this interaction in the context of inner ear development and deafness remains elusive.
Chd8 has been found in the proteome of HCs from P4–P7 organs of Corti derived from a
Pou4f3-eGFP transgenic mouse model
[41][49]. It has been suggested as a candidate deafness gene (DFNA53) with a possible functional role in HCs. In another study, RNA-seq data showed that CHD8 knock-out hES cell-derived neuroectodermal cells are enriched in genes for “inner ear morphogenesis” gene-ontology terms such as
PAX8,
FGF9 and
MYO15A [42][50]. This observed increase in “inner ear” genes might arise since CHD8 has been shown to interact directly with β-catenin and be recruited to the promoter regions of numerous β-catenin-responsive genes acting as a repressor of both β-catenin and the Wnt pathway
[43][44][51,52]. Several identified genes (
PAX8, DLX6, FGF9) are upstream or downstream effectors of β-catenin/Wnt pathway modulation
[45][46][53,54].
In a recent study, the deletion of Chd8 in mice under the control of the Atoh1 promoter—expressed explicitly in HCs and spiral ganglion neurons—did not affect ABR
[47][55]. Moreover, the development of HCs and auditory neurons seemed normal. Therefore, Chd8 does not appear to be necessary for cochlea development and hearing function in mice, while it seems essential in humans. The functional role of CHD8 in inner ear development and hearing loss remains to be established.