The highly conserved and dynamically reversible N6-methyladenine (m6A) modification has emerged as a critical gene expression regulator by affecting RNA splicing, translation efficiency, and stability at the post-transcriptional level, which has been established to be involved in various physiological and pathological processes, including glycolipid metabolism and the development of glycolipid metabolic disease (GLMD). Hence, accumulating studies have focused on the effects and regulatory mechanisms of m6A modification on glucose metabolism, lipid metabolism, and GLMD. Glucose metabolism involves a very complex regulatory network, including anaerobic glycolysis, aerobic oxidation, pentose phosphate pathway, glycogen synthesis, and gluconeogenesis. An increasing number of studies have reported that m6A modification is an important regulatory mechanism of glucose homeostasis and downstream effects.
- N6-methyladenosine methylation
- glucose metabolism
- lipid metabolism
- glycolipid metabolic disease
1. Introduction
2. m6A Methylation
M6A mainly refers to the methylation of the sixth nitrogen of adenosine, which accounts for the most abundant internal modification that occurs in eukaryotic mRNA. Other than mRNA, m6A occurs in long non-coding RNA (lncRNA), circular RNA (circRNA), and microRNA (miRNA) [13,14,15][13][14][15]. The highly conserved and dynamic m6A modification level has been established to be regulated by methyltransferases (termed “writers”) and demethylases (termed “erasers”) (Table 1). The m6A writers comprised Methyltransferase-like protein 3 (METTL3), methyltransferase-like protein 14 (METTL14) [16], methyltransferase-like protein 16 (METTL16), Wilms tumor 1-associated protein (WTAP), Vir-like m6A methyltransferase associated (VIRMA/KIAA1429), RNA-binding motifs protein15/15B (RBM15/15B), and Zinc Finger CCCH-Type Containing 13 (ZC3H13). Typically, METTL3, METTL14, and WTAP form a methyltransferase complex (MTC), which recognizes a consensus RNA sequence RRACH (R = G or A; H = A, C or U) and catalyzes the m6A modification on mRNAs, while METTL16 regulates S-adenosylmethionine (SAM) homeostasis [17]. VIRMA recruits MTC to specific mRNA regions and interacts with cleavage and polyadenylation specific factor 5/6 (CPSF5/6) [18]. RBM15/15B mainly guides METTL3-METTL14 heterodimer to uracil U-rich RNA sites for methylation [19]. ZC3H13 bridges WTAP to the mRNA binding factor Nito and contributes to the nuclear localization of MTC [20]. Additionally, IME4 and MUM2 mediate m6A modification of yeast mRNA [21].| Type | Regulator | Biological Function | References |
|---|---|---|---|
| m6A writers | METTL3 | Catalyzes m6A modification | [16] |
| METTL14 | Assists METTL3 to recognize the specific subtract and enhances the stability of MTC structure | [16] | |
| METTL16 | Catalyzes m6A modification and regulates SAM homeostasis | [17] | |
| WTAP | Promotes the cellular m6A deposition | [18] | |
| VIRMA (KIAA1429) |
Guides the core component of MTC to specific RNA region and interacts with cleavage and polyadenylation specific factor 5/6 (CPSF5/6) | [18] | |
| RBM15/15B | Recruits METTL3-METTL14 heterodimer to specific RNA sites | [19] | |
| ZC3H13 | Contributes to the nuclear localization of MTC | [20] | |
| IME4 | Mediates m6A modification of yeast mRNA | [21] | |
| MUM2 | Mediates m6A modification of yeast mRNA | [21] | |
| m6A erasers | FTO | Removes m6A modification to promote mRNA splicing and translation | [22] |
| ALKBH5 | Removes m6A modification to promote mRNA splicing, mRNA nuclear output, and long 3′-UTR mRNA production | [23] | |
| m6A readers | YTHDF1 | Promotes mRNA translation and protein synthesis | [24] |
| YTHDF2 | Reduces mRNA stability and regulates mRNA localization | [24] | |
| YTHDF3 | Interacts with YTHDF1 to promote mRNA translation or assists YTHDF2-mediated RNA degradation | [24] | |
| YTHDC1 | Contributes to RNA splicing, export, and transcriptional silencing | [25] | |
| YTHDC2 | Promotes the translation of target RNA but reduces their abundance | [26] | |
| eIF3 | Promotes mRNA translation | [27] | |
| IGF2BPs | Enhances the stability and translation of target RNA | [28] | |
| HNRNPA2B1 | Regulates alternative splicing and primary microRNA processing | [29] | |
| HNRNPC/G | Mediates mRNA splicing | [29] |
3. m6A Modification and Glucose Metabolism
Glucose metabolism involves a very complex regulatory network, including anaerobic glycolysis, aerobic oxidation, pentose phosphate pathway, glycogen synthesis, and gluconeogenesis [30]. An increasing number of studies have reported that m6A modification is an important regulatory mechanism of glucose homeostasis and downstream effects (Figure 1).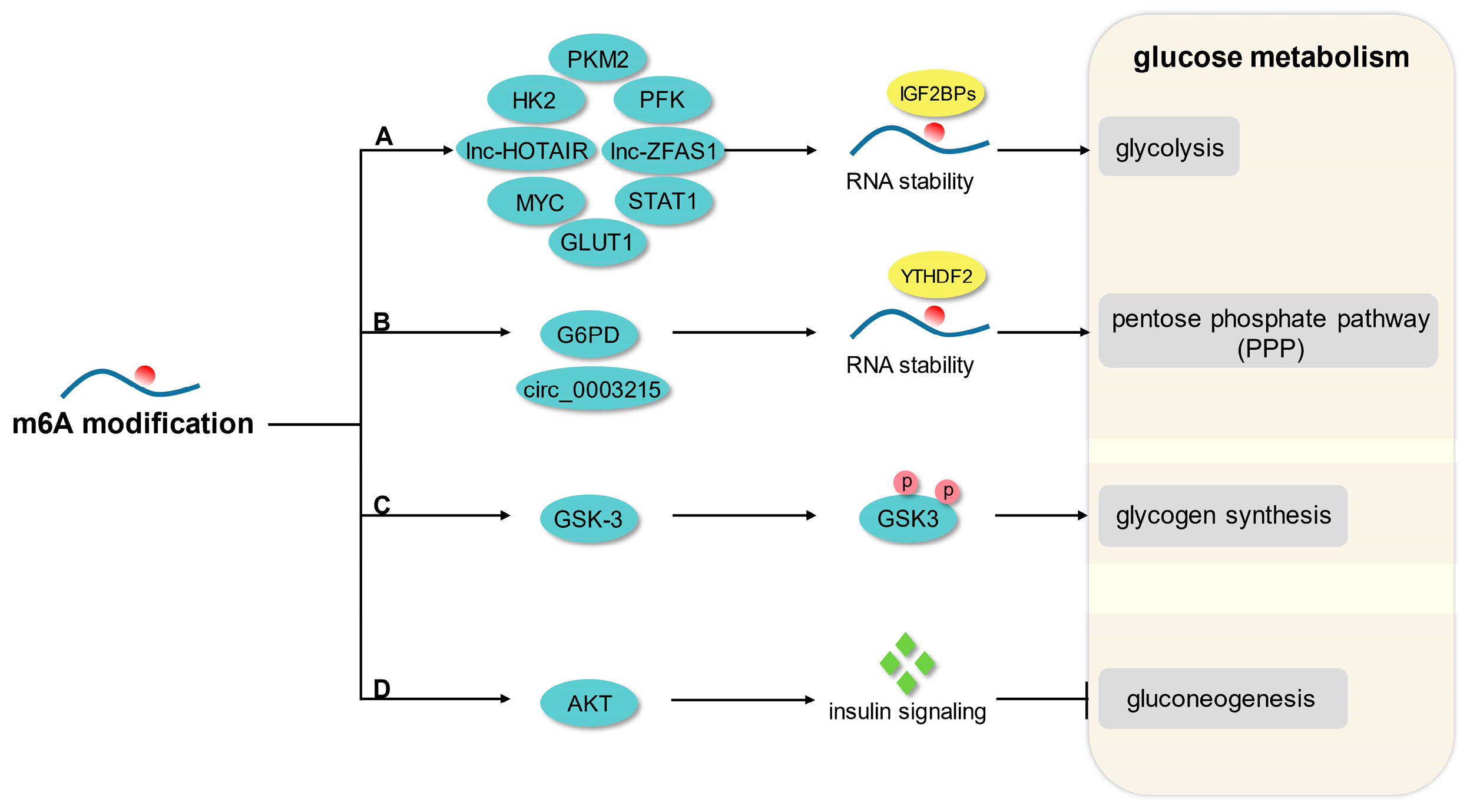
3.1. Glycolysis
Glycolysis is a pivotal energy-producing pathway in organisms, which decomposes glucose to pyruvate under anaerobic conditions, with the release of free energy into adenosine triphosphate (ATP). Typically, monosaccharides are transported to the cytoplasm by glucose transporters (GLUTs) or sodium-dependent glucose cotransporters (SGLTs), then undergo the preparation phase of glucose activation and cleavage and the release energy phase of oxidative phosphorylation, which are closely related to hexokinase (HK), phosphofructokinase-1 (PFK-1) and pyruvate kinase (PK), and ultimately generate pyruvate and ATP [31]. A growing number of studies have uncovered the broad effects of m6A modification on metabolic networks by regulating glycolytic genes and signaling pathways. METTL3/IGF2BP2-mediated m6A modification has been reported to promote glycolysis by enhancing the stability of HK2 and GLUT1 [32,33][32][33]. METTL14-mediated m6A modification not only promotes glycolysis by attenuating sirtuin6 (SIRT6) stability but also facilitates hypoxia-inducible factor 1 subunit alpha (HIF1A)-mediated glycolysis and cell proliferation by inhibiting the expression of phosphatase LHPP [34,35][34][35]. WTAP enhances glycolytic activity by mediating m6A methylation of HK2 and enolase 1 (ENO1) mRNA [36,37,38][36][37][38]. The m6A methyltransferase KIAA1429 positively regulates aerobic glycolysis in a GLUTI or HK2-dependent manner [39[39][40],40], and ZC3H13 facilitates glycolysis by enhancing the stability of pyruvate kinase M2 (PKM2) mRNA [41]. In addition, FTO-mediated demethylation promotes HK2 and PKM2-mediated glycolysis via upregulating the expression of lncRNA HOTAIR and autophagy associated 5 gene (ATG5), respectively [42[42][43],43], while down-regulation of FTO participates MYC-mediated cellular glycolysis and the regulation of IL-6/JAK2/STAT3 signaling pathways [44,45][44][45]. FTO/YTHDF2 mediates post-transcriptional upregulation of phosphofructokinase platelet (PFKP) and lactate dehydrogenase B (LDHB) and activates aerobic glycolysis [46]. ALKBH5 is involved in the regulation of casein kinase 2 (CK2) α-mediated glycolysis in an m6A-dependent manner [47]. M6A reader IMP2 enhances the stability of lncRNA ZFAS1 and facilitates the exposure of ATP-binding sites, thereby accelerating ATP hydrolysis and glycolysis [48]. IGF2BP1/2 is involved in the regulation of MYC-mediated glycolysis and provides an additional energy source for cell metabolism [49,50,51][49][50][51]. In cellular metabolism, pyruvate dehydrogenase kinase 4 (PDK4) methylation can be recognized by YTHDF1/eEF-2 complex and IGF2BP3 to direct carbon flux from oxidative phosphorylation (OXPHOS) to glycolysis [52]. The interaction of YTHDF2 with RNA-binding motif protein 4 (RBM4) promotes signal transduction and activator of transcription 1 (STAT1)-mediated glycolysis, which participates in regulating macrophage polarization and inflammatory factor expression [53].3.2. Pentose Phosphate Pathway
The pentose phosphate pathway (PPP), also known as the hexose phosphate bypass, is generally divided into two branches: oxidative and non-oxidative. During the highly active oxidation phase in most eukaryotes, glucose-6-phosphate (G-6-P) is converted to ribulose-5-phosphate, carbon dioxide, and nicotinamide adenine dinucleotide phosphate (NADPH) [54]. The non-oxidative branch is nearly ubiquitous and supports the nucleic acid skeleton and aromatic amino acid biosynthesis by increasing the expression of 5-phosphate ribose and erythritol-4-phosphate [55,56][55][56]. As a key enzyme of PPP, glucose-6-phosphate dehydrogenase (G6PD) is overactive in metabolic pathways and participates in the regulation of redox status. Recent studies have demonstrated that tumor cells respond to chemotherapy-induced reactive oxygen species (ROS) accumulation by activating PPP to increase NADPH, thereby adapting to oxidative stress and maintaining malignant cell proliferation [57]. In a glioma, ALKBH5 enhances the stability of G6PD mRNA and PPP flux by eliminating m6A methylation [58]. YTHDF2 facilitates mRNA translation of G6PD in an m6A-dependent manner, thereby enhancing PPP activity and tumor cell viability [59]. In addition, YTHDF2 promotes the miR-663b/DLG4/G6PD axis and pentose phosphate pathway by mediating circ_0003215 RNA degradation [60].3.3. Glycogen Synthesis and Gluconeogenesis
As a storage form of sugar, glycogen synthesis refers to the process of converting activated glucose into glycogen under the catalysis of glycogen synthase. Glycogen synthase activity is regulated by phosphorylation/de-phosphorylation of various serine/threonine kinases, among which glycogen synthase kinase 3 (GSK-3) is widely involved in the physiological and metabolic processes. Under normal feeding conditions, enhanced insulin signaling activates protein kinase B (AKT), which then inactivates GSK-3 through phosphorylation and ultimately promotes glycogen synthesis in response to increased glucose uptake. In contrast, fasting activates GSK-3 by de-phosphorylation, which inhibits glycogen synthesis and facilitates glycogenolysis, supplying the body with fuel reserve [61]. As a highly conserved negative regulator of receptor tyrosine kinases, cytokines, and Wnt signaling pathways, GSK-3 participates in the m6A methylation regulatory network and affects multiple downstream effectors. Similarly, downregulated microRNA-6125 and hypoxia-induced lncRNA STEAP3-AS1 interact competitively with YTHDF2, which results in phosphorylation and inactivation of GSK3β, activation of Wnt/β-catenin signaling pathways, and glycogen synthesis [62,63][62][63]. METTL14 negatively regulates the expression of fibroblast growth factor receptor 4 (FGFR4) in an m6A-dependent manner, while FGFR4 activates glycogen synthesis and the β-catenin/TCF-4 signaling pathway through phosphorylation of GSK-3β [64]. M6A mediates phosphorylation of AKT/GSK-3β and activation of tensin homolog protein (PTEN), which promotes glycogen synthesis and protects neurons from pyroptosis induced by cerebral ischemia/reperfusion (I/R) [65]. Cardiac hypertrophy-associated PIWI-interacting RNA (CHAPIR) competitively binds METTL3 and blocks m6A methylation of polymerase family member 10 (PARP10), while up-regulation of PARP10 facilitates glycogen synthesis and pathological cardiac hypertrophy by inhibiting the kinase activity of GSK-3β [66]. The process by which organisms synthesize glucose or glycogen from non-sugar precursors such as lactic acid, glycerol, and glycogenic amino acids is known as gluconeogenesis. Under starvation, the liver promotes gluconeogenesis by decreasing insulin concentration and increasing glucagon concentration, which is the main cause of diabetic hyperglycemic phenotype [67]. METTL14 deficiency leads to decreased β-cell mass and insulin secretion, but β-cell-specific knockdown of METTL14 enhances insulin signaling and reduces hepatic gluconeogenesis under a high-fat diet (HFD), thus improving insulin sensitivity with compensated [68]. This suggests that m6A methylation regulates the gluconeogenic flux in response to insulin signaling, which is of great significance for glucose homeostasis.References
- Jia, G.; Fu, Y.; Zhao, X.; Dai, Q.; Zheng, G.; Yang, Y.; Yi, C.; Lindahl, T.; Pan, T.; Yang, Y.G.; et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat. Chem. Biol. 2011, 7, 885–887.
- Zheng, G.; Dahl, J.A.; Niu, Y.; Fedorcsak, P.; Huang, C.M.; Li, C.J.; Vagbo, C.B.; Shi, Y.; Wang, W.L.; Song, S.H.; et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol. Cell 2013, 49, 18–29.
- Wei, C.; Gershowitz, A.; Moss, B. Methylated nucleotides block 5′ terminus of HeLa cell messenger RNA. Cell 1975, 4, 379–386.
- Bokar, J.; Shambaugh, M.; Polayes, D.; Matera, A.; Rottman, F. Purification and cDNA cloning of the AdoMet-binding subunit of the human mRNA (N6-adenosine)-methyltransferase. RNA 1997, 3, 1233–1247.
- He, L.; Li, H.; Wu, A.; Peng, Y.; Shu, G.; Yin, G. Functions of N6-methyladenosine and its role in cancer. Mol. Cancer 2019, 18, 176.
- Huang, H.; Weng, H.; Chen, J. m6A Modification in Coding and Non-coding RNAs: Roles and Therapeutic Implications in Cancer. Cancer Cell 2020, 37, 270–288.
- Li, A.; Chen, Y.-S.; Ping, X.-L.; Yang, X.; Xiao, W.; Yang, Y.; Sun, H.-Y.; Zhu, Q.; Baidya, P.; Wang, X.; et al. Cytoplasmic m6A reader YTHDF3 promotes mRNA translation. Cell Res. 2017, 27, 444–447.
- Śledź, P.; Jinek, M. Structural insights into the molecular mechanism of the m6A writer complex. eLife 2016, 5, e18434.
- Wang, X.; Lu, Z.; Gomez, A.; Hon, G.C.; Yue, Y.; Han, D.; Fu, Y.; Parisien, M.; Dai, Q.; Jia, G.; et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature 2013, 505, 117–120.
- Xiao, W.; Adhikari, S.; Dahal, U.; Chen, Y.-S.; Hao, Y.-J.; Sun, B.-F.; Sun, H.-Y.; Li, A.; Ping, X.-L.; Lai, W.-Y.; et al. Nuclear m6A Reader YTHDC1 Regulates mRNA Splicing. Mol. Cell 2016, 61, 507–519.
- Cai, Z.; Deng, X.; Zhao, L.; Wang, X.; Yang, L.; Yuan, G. The relationship between Schistosoma and glycolipid metabolism. Microb. Pathog. 2021, 159, 105120.
- Wu, J.; Frazier, K.; Zhang, J.; Gan, Z.; Wang, T.; Zhong, X. Emerging role of m6A RNA methylation in nutritional physiology and metabolism. Obes. Rev. 2019, 21, e12942.
- Dominissini, D.; Moshitch-Moshkovitz, S.; Schwartz, S.; Salmon-Divon, M.; Ungar, L.; Osenberg, S.; Cesarkas, K.; Jacob-Hirsch, J.; Amariglio, N.; Kupiec, M.; et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 2012, 485, 201–206.
- Meyer, K.D.; Saletore, Y.; Zumbo, P.; Elemento, O.; Mason, C.E.; Jaffrey, S.R. Comprehensive Analysis of mRNA Methylation Reveals Enrichment in 3′ UTRs and near Stop Codons. Cell 2012, 149, 1635–1646.
- Yang, Y.; Fan, X.; Mao, M.; Song, X.; Wu, P.; Zhang, Y.; Jin, Y.; Yang, Y.; Chen, L.-L.; Wang, Y.; et al. Extensive translation of circular RNAs driven by N6-methyladenosine. Cell Res. 2017, 27, 626–641.
- Wang, P.; Doxtader, K.A.; Nam, Y. Structural Basis for Cooperative Function of Mettl3 and Mettl14 Methyltransferases. Mol. Cell 2016, 63, 306–317.
- Zhou, K.I.; Pan, T. Structures of the m6A Methyltransferase Complex: Two Subunits with Distinct but Coordinated Roles. Mol. Cell 2016, 63, 183–185.
- Yue, Y.; Liu, J.; Cui, X.; Cao, J.; Luo, G.; Zhang, Z.; Cheng, T.; Gao, M.; Shu, X.; Ma, H.; et al. VIRMA mediates preferential m6A mRNA methylation in 3′UTR and near stop codon and associates with alternative polyadenylation. Cell Discov. 2018, 4, 10.
- Patil, D.P.; Chen, C.-K.; Pickering, B.F.; Chow, A.; Jackson, C.; Guttman, M.; Jaffrey, S.R. m6A RNA methylation promotes XIST-mediated transcriptional repression. Nature 2016, 537, 369–373.
- Wen, J.; Lv, R.; Ma, H.; Shen, H.; He, C.; Wang, J.; Jiao, F.; Liu, H.; Yang, P.; Tan, L.; et al. Zc3h13 Regulates Nuclear RNA m6A Methylation and Mouse Embryonic Stem Cell Self-Renewal. Mol. Cell 2018, 69, 1028–1038.e6.
- Schwartz, S.; Agarwala, S.D.; Mumbach, M.; Jovanovic, M.; Mertins, P.; Shishkin, A.; Tabach, Y.; Mikkelsen, T.S.; Satija, R.; Ruvkun, G.; et al. High-Resolution Mapping Reveals a Conserved, Widespread, Dynamic mRNA Methylation Program in Yeast Meiosis. Cell 2013, 155, 1409–1421.
- Wei, J.; Liu, F.; Lu, Z.; Fei, Q.; Ai, Y.; He, P.C.; Shi, H.; Cui, X.; Su, R.; Klungland, A.; et al. Differential m6A, m6Am, and m1A Demethylation Mediated by FTO in the Cell Nucleus and Cytoplasm. Mol. Cell 2018, 71, 973–985.e5.
- Tang, C.; Klukovich, R.; Peng, H.; Wang, Z.; Yu, T.; Zhang, Y.; Zheng, H.; Klungland, A.; Yan, W. ALKBH5-dependent m6A demethylation controls splicing and stability of long 3′-UTR mRNAs in male germ cells. Proc. Natl. Acad. Sci. USA 2017, 115, E325–E333.
- Wang, X.; Zhao, B.S.; Roundtree, I.A.; Lu, Z.; Han, D.; Ma, H.; Weng, X.; Chen, K.; Shi, H.; He, C. N6-methyladenosine Modulates Messenger RNA Translation Efficiency. Cell 2015, 161, 1388–1399.
- Roundtree, I.A.; Luo, G.-Z.; Zhang, Z.; Wang, X.; Zhou, T.; Cui, Y.; Sha, J.; Huang, X.; Guerrero, L.; Xie, P.; et al. YTHDC1 mediates nuclear export of N6-methyladenosine methylated mRNAs. eLife 2017, 6, e31311.
- Hsu, P.J.; Zhu, Y.; Ma, H.; Guo, Y.; Shi, X.; Liu, Y.; Qi, M.; Lu, Z.; Shi, H.; Wang, J.; et al. Ythdc2 is an N6-methyladenosine binding protein that regulates mammalian spermatogenesis. Cell Res. 2017, 27, 1115–1127.
- Meyer, K.D.; Patil, D.P.; Zhou, J.; Zinoviev, A.; Skabkin, M.A.; Elemento, O.; Pestova, T.V.; Qian, S.-B.; Jaffrey, S.R. 5′ UTR m6A Promotes Cap-Independent Translation. Cell 2015, 163, 999–1010.
- Huang, H.; Weng, H.; Sun, W.; Qin, X.; Shi, H.; Wu, H.; Zhao, B.S.; Mesquita, A.; Liu, C.; Yuan, C.L.; et al. Recognition of RNA N6-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat. Cell Biol. 2018, 20, 285–295, Correction in Nat. Cell Biol. 2018, 20, 1098; Correction in Nat. Cell Biol. 2020, 22, 1288.
- Wu, B.; Su, S.; Patil, D.P.; Liu, H.; Gan, J.; Jaffrey, S.R.; Ma, J. Molecular basis for the specific and multivariant recognitions of RNA substrates by human hnRNP A2/B1. Nat. Commun. 2018, 9, 420.
- Boucheé, C.; Serdy, S.; Kahn, C.R.; Goldfine, A. The Cellular Fate of Glucose and Its Relevance in Type 2 Diabetes. Endocr. Rev. 2004, 25, 807–830.
- Butterfield, D.A.; Halliwell, B. Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat. Rev. Neurosci. 2019, 20, 148–160.
- Shen, C.; Xuan, B.; Yan, T.; Ma, Y.; Xu, P.; Tian, X.; Zhang, X.; Cao, Y.; Ma, D.; Zhu, X.; et al. m6A-dependent glycolysis enhances colorectal cancer progression. Mol. Cancer 2020, 19, 72.
- Zheng, Y.; Wang, Y.; Liu, Y.; Xie, L.; Ge, J.; Yu, G.; Zhao, G. N6-Methyladenosine Modification of PTTG3P Contributes to Colorectal Cancer Proliferation via YAP1. Front. Oncol. 2021, 11, 669731.
- Du, L.; Li, Y.; Kang, M.; Feng, M.; Ren, Y.; Dai, H.; Wang, Y.; Wang, Y.; Tang, B. USP48 Is Upregulated by Mettl14 to Attenuate Hepatocellular Carcinoma via Regulating SIRT6 Stabilization. Cancer Res. 2021, 81, 3822–3834.
- Lin, J.-X.; Lian, N.-Z.; Gao, Y.-X.; Zheng, Q.-L.; Yang, Y.-H.; Ma, Y.-B.; Xiu, Z.-S.; Qiu, Q.-Z.; Wang, H.-G.; Zheng, C.-H.; et al. m6A methylation mediates LHPP acetylation as a tumour aerobic glycolysis suppressor to improve the prognosis of gastric cancer. Cell Death Dis. 2022, 13, 463.
- Lyu, Y.; Zhang, Y.; Wang, Y.; Luo, Y.; Ding, H.; Li, P.; Ni, G. HIF-1α Regulated WTAP Overexpression Promoting the Warburg Effect of Ovarian Cancer by m6A-Dependent Manner. J. Immunol. Res. 2022, 2022, 6130806.
- Ou, B.; Liu, Y.; Yang, X.; Xu, X.; Yan, Y.; Zhang, J. C5aR1-positive neutrophils promote breast cancer glycolysis through WTAP-dependent m6A methylation of ENO1. Cell Death Dis. 2021, 12, 737.
- Yu, H.; Zhao, K.; Zeng, H.; Li, Z.; Chen, K.; Zhang, Z.; Li, E.; Wu, Z. N6-methyladenosine (m6A) methyltransferase WTAP accelerates the Warburg effect of gastric cancer through regulating HK2 stability. Biomed. Pharmacother. 2020, 133, 111075.
- Li, Y.; He, L.; Wang, Y.; Tan, Y.; Zhang, F. N6-methyladenosine methyltransferase KIAA1429 elevates colorectal cancer aerobic glycolysis via HK2-dependent manner. Bioengineered 2022, 13, 11923–11932.
- Yang, D.; Chang, S.; Li, F.; Ma, M.; Yang, J.; Lv, X.; Huangfu, L.; Jia, C. m6A transferase KIAA1429-stabilized LINC00958 accelerates gastric cancer aerobic glycolysis through targeting GLUT1. IUBMB Life 2021, 73, 1325–1333.
- Wang, Q.; Xie, H.; Peng, H.; Yan, J.; Han, L.; Ye, G. ZC3H13 Inhibits the Progression of Hepatocellular Carcinoma through m6A-PKM2-Mediated Glycolysis and Enhances Chemosensitivity. J. Oncol. 2021, 2021, 1328444.
- Sun, X.; Li, Q.; Yang, L. Sevoflurane Inhibits lncRNA HOTAIR-Modulated Stability of HK2 mRNA in a m6A-Dependent Manner to Dampen Aerobic Glycolysis and Proliferation in Lung Cancer. BioMed. Res. Int. 2022, 2022, 4668774.
- Wang, H.; Liang, Z.; Gou, Y.; Li, Z.; Cao, Y.; Jiao, N.; Tan, J.; Yu, Y.; Zhang, Z. FTO-dependent N(6)-Methyladenosine regulates the progression of endometriosis via the ATG5/PKM2 Axis. Cell. Signal. 2022, 98, 110406.
- Huang, J.; Sun, W.; Wang, Z.; Lv, C.; Zhang, T.; Zhang, D.; Dong, W.; Shao, L.; He, L.; Ji, X.; et al. FTO suppresses glycolysis and growth of papillary thyroid cancer via decreasing stability of APOE mRNA in an N6-methyladenosine-dependent manner. J. Exp. Clin. Cancer Res. 2022, 41, 42.
- Yang, X.; Shao, F.; Guo, D.; Wang, W.; Wang, J.; Zhu, R.; Gao, Y.; He, J.; Lu, Z. WNT/β-catenin-suppressed FTO expression increases m6A of c-Myc mRNA to promote tumor cell glycolysis and tumorigenesis. Cell Death Dis. 2021, 12, 462.
- Qing, Y.; Dong, L.; Gao, L.; Li, C.; Li, Y.; Han, L.; Prince, E.; Tan, B.; Deng, X.; Wetzel, C.; et al. R-2-hydroxyglutarate attenuates aerobic glycolysis in leukemia by targeting the FTO/m6A/PFKP/LDHB axis. Mol. Cell 2021, 81, 922–939.e9.
- Yu, H.; Yang, X.; Tang, J.; Si, S.; Zhou, Z.; Lu, J.; Han, J.; Yuan, B.; Wu, Q.; Lu, Q.; et al. ALKBH5 Inhibited Cell Proliferation and Sensitized Bladder Cancer Cells to Cisplatin by m6A-CK2α-Mediated Glycolysis. Mol. Ther. Nucleic. Acids 2020, 23, 27–41.
- Lu, S.; Han, L.; Hu, X.; Sun, T.; Xu, D.; Li, Y.; Chen, Q.; Yao, W.; He, M.; Wang, Z.; et al. N6-methyladenosine reader IMP2 stabilizes the ZFAS1/OLA1 axis and activates the Warburg effect: Implication in colorectal cancer. J. Hematol. Oncol. 2021, 14, 188.
- Hu, C.; Liu, T.; Han, C.; Xuan, Y.; Jiang, D.; Sun, Y.; Zhang, X.; Zhang, W.; Xu, Y.; Liu, Y.; et al. HPV E6/E7 promotes aerobic glycolysis in cervical cancer by regulating IGF2BP2 to stabilize m6A-MYC expression. Int. J. Biol. Sci. 2022, 18, 507–521.
- Luo, F.; Lin, K. N6-methyladenosine (m6A) reader IGF2BP1 accelerates gastric cancer aerobic glycolysis in c-Myc-dependent manner. Exp. Cell Res. 2022, 417, 113176.
- Wang, Y.; Lu, J.-H.; Wu, Q.-N.; Jin, Y.; Wang, D.-S.; Chen, Y.-X.; Liu, J.; Luo, X.-J.; Meng, Q.; Pu, H.-Y.; et al. LncRNA LINRIS stabilizes IGF2BP2 and promotes the aerobic glycolysis in colorectal cancer. Mol. Cancer 2019, 18, 174.
- Li, Z.; Peng, Y.; Li, J.; Chen, Z.; Chen, F.; Tu, J.; Lin, S.; Wang, H. N6-methyladenosine regulates glycolysis of cancer cells through PDK4. Nat. Commun. 2020, 11, 2578.
- Huangfu, N.; Zheng, W.; Xu, Z.; Wang, S.; Wang, Y.; Cheng, J.; Li, Z.; Cheng, K.; Zhang, S.; Chen, X.; et al. RBM4 regulates M1 macrophages polarization through targeting STAT1-mediated glycolysis. Int. Immunopharmacol. 2020, 83, 106432.
- Stincone, A.; Prigione, A.; Cramer, T.; Wamelink, M.M.C.; Campbell, K.; Cheung, E.; Olin-Sandoval, V.; Grüning, N.-M.; Krüger, A.; Tauqeer Alam, M.; et al. The return of metabolism: Biochemistry and physiology of the pentose phosphate pathway. Biol. Rev. 2015, 90, 927–963.
- Clasquin, M.F.; Melamud, E.; Singer, A.; Gooding, J.R.; Xu, X.; Dong, A.; Cui, H.; Campagna, S.R.; Savchenko, A.; Yakunin, A.F.; et al. Riboneogenesis in Yeast. Cell 2011, 145, 969–980.
- Wang, L.; Xie, J.; Schultz, P.G. Expanding the Genetic Code. Annu. Rev. Biophys. Biomol. Struct. 2006, 35, 225–249.
- Hayes, J.D.; Dinkova-Kostova, A.T.; Tew, K.D. Oxidative Stress in Cancer. Cancer Cell 2020, 38, 167–197.
- Liu, Z.; Chen, Y.; Wang, L.; Ji, S. ALKBH5 Promotes the Proliferation of Glioma Cells via Enhancing the mRNA Stability of G6PD. Neurochem. Res. 2021, 46, 3003–3011.
- Sheng, H.; Li, Z.; Su, S.; Sun, W.; Zhang, X.; Li, L.; Li, J.; Liu, S.; Lu, B.; Zhang, S.; et al. YTH domain family 2 promotes lung cancer cell growth by facilitating 6-phosphogluconate dehydrogenase mRNA translation. Carcinog. 2019, 41, 541–550.
- Chen, B.; Hong, Y.; Gui, R.; Zheng, H.; Tian, S.; Zhai, X.; Xie, X.; Chen, Q.; Qian, Q.; Ren, X.; et al. N6-methyladenosine modification of circ_0003215 suppresses the pentose phosphate pathway and malignancy of colorectal cancer through the miR-663b/DLG4/G6PD axis. Cell Death Dis. 2022, 13, 804.
- Han, H.-S.; Kang, G.; Kim, J.S.; Choi, B.H.; Koo, S.-H. Regulation of glucose metabolism from a liver-centric perspective. Exp. Mol. Med. 2016, 48, e218.
- Li, H.; Zhang, N.; Jiao, X.; Wang, C.; Sun, W.; He, Y.; Ren, G.; Huang, S.; Li, M.; Chang, Y.; et al. Downregulation of microRNA-6125 promotes colorectal cancer growth through YTHDF2-dependent recognition of N6-methyladenosine-modified GSK3β. Clin. Transl. Med. 2021, 11, e602.
- Zhou, L.; Jiang, J.; Huang, Z.; Jin, P.; Peng, L.; Luo, M.; Zhang, Z.; Chen, Y.; Na Xie, N.; Gao, W.; et al. Hypoxia-induced lncRNA STEAP3-AS1 activates Wnt/β-catenin signaling to promote colorectal cancer progression by preventing m6A-mediated degradation of STEAP3 mRNA. Mol. Cancer 2022, 21, 168.
- Zou, Y.; Zheng, S.; Xie, X.; Ye, F.; Hu, X.; Tian, Z.; Yan, S.-M.; Yang, L.; Kong, Y.; Tang, Y.; et al. N6-methyladenosine regulated FGFR4 attenuates ferroptotic cell death in recalcitrant HER2-positive breast cancer. Nat. Commun. 2022, 13, 2672.
- Diao, M.-Y.; Zhu, Y.; Yang, J.; Xi, S.-S.; Wen, X.; Gu, Q.; Hu, W. Hypothermia protects neurons against ischemia/reperfusion-induced pyroptosis via m6A-mediated activation of PTEN and the PI3K/Akt/GSK-3β signaling pathway. Brain Res. Bull. 2020, 159, 25–31.
- Gao, X.-Q.; Zhang, Y.-H.; Liu, F.; Ponnusamy, M.; Zhao, X.-M.; Zhou, L.-Y.; Zhai, M.; Liu, C.-Y.; Li, X.-M.; Wang, M.; et al. The piRNA CHAPIR regulates cardiac hypertrophy by controlling METTL3-dependent N6-methyladenosine methylation of Parp10 mRNA. Nature 2020, 22, 1319–1331.
- Hatting, M.; Tavares, C.D.; Sharabi, K.; Rines, A.K.; Puigserver, P. Insulin regulation of gluconeogenesis. Ann. N. Y. Acad. Sci. 2017, 1411, 21–35.
- Liu, J.; Luo, G.; Sun, J.; Men, L.; Ye, H.; He, C.; Ren, D. METTL14 is essential for β-cell survival and insulin secretion. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2019, 1865, 2138–2148.