Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Jessie Wu and Version 3 by Jessie Wu.
Endocytosis is a mechanistic process, associated with internalization of the extracellular materials such as microbes, cellular components, nutrients, or macromolecules. Conventionally, eukaryotic cells use the endocytosis process for the absorption of molecules and secretion of signaling transmitters (hormones and cytokines) to maintain cellular homeostasis. Endocytosis machinery is a well-conserved physiological process in lower to higher organisms, which has been frequently acquired for cellular defense, immune responses, uptake, and energy metabolism.
- endocytosis
- pinocytosis
- diseases
- Cancer
1. Cancer
Endocytosis is a complex cellular event involved in homeostasis and communication to extracellular milieu through internalization of the plasma membrane along with its integral membrane proteins, immunoglobulins, receptors and their ligands, and nutrients. It is a crucial signaling event that plays a key role in cell cycle regulation, mitosis, and apoptosis. Endocytosis is a regulated signaling mechanism and plays a potential role in tumor suppressor pathways. It plays a critical role in signaling through endosomes and rescue degradation of signaling molecules involved in cancer signaling, thus it appears as a potential target in oncogenic pathways. Further, endocytosis is involved in activation of certain cancer receptors such as Epidermal growth factor receptor (EGFR), Transferrin receptor (TfR), and Notch receptor [1]. In addition, in human tumors, altered expression of various endocytic regulatory factors such as clathrin-hc, clathrin- like, Caveolin-1, Nexin-1, and Numb along with driver mutations are crucial for endocytosis [2][3]. Endocytic protein Numb governs the level and activity of tumor suppressive protein p53. Numb inhibits the degradation of p53 by forming the tricomplex of p53 and Hdm2 where it suppresses the ligase activity of Hdm2 [4]. Thus, perturbation in Numb levels may alter the expression of the p53-associated cellular process, such as response to DNA damage, induction of checkpoint, and apoptosis-associated proteins [5]. Furthermore, a remarkable decline in the levels of Numb expression has been observed in approximately 50% of breast cancer [4]. The disruption of Numb expression might have a severe impact on tumorigenesis. The endocytic activity of Numb is associated to Numb–Notch interactions for cell proliferation and differentiation. Importantly, endocytosis plays an indispensable role in the aggressive nature of cancer. Endocytosis appears as an important regulator of tumor metastasis [6]. In cancer, deregulation of several endocytic proteins is involved in migration and invasion. There are some metastatic suppressor genes such as Kisspeptin-1 (KISS1), and metastasis suppressor protein 1 (MTSS1) whose activity depends on alteration in the endocytosis process [6]. Kisspeptin-1 (KISS-1) inhibits cell motility, proliferation, invasion, and metastasis in cancers [7]. However, in breast cancer, it induces invasion. MTSS1 acts as a scaffold protein and inhibits the metastasis in various cancers. However, in head and neck squamous cell carcinoma, a low level of MTSS1 augment the EGF signaling and induce cell proliferation [6]. In contrast, a high level of MTSS1 exhibits a negative influence on EGF signaling and triggers metastasis [8].
In addition, several studies showed the involvement of different classes of endocytic proteins in the invasion of multiple cancer types, including colon, breast, colorectal, and non-small cell lung carcinoma (NSCLC). Caveolin endocytic protein shows a regulatory function for breast, prostate, and ovarian cancer. In the early stage of these cancers, caveolin acts as a tumor suppressor, while in advanced stage, it is associated with tumor progression and metastasis [9][10]. Clathrin-mediated endocytic protein AP2 modulates the cell migration and invasion of pancreatic, ovarian, and melanoma cancer through CXCR2 [11]. Endosomal trafficking proteins such as ARF1 regulate breast cancer cell proliferation and migration through regulating the interaction of β1 integrin and protein of focal adhesion (paxillin, Fak, talin) [12][13]. ARF6 promotes cellular motility and invasion of glioma and breast cancer cells by inducing internalization of E-cadherin and breakdown of adherence junction [14]. Further, endocytic proteins of the RAB subfamily such as RAB3C and RAB3D control invasion and metastasis of colorectal and breast cancer, respectively. Elevated expression of RAB3C promotes in vivo migration, invasion, and metastasis of colorectal cancer while RAB3D induced breast cancer cell invasion by activating Akt/GSK-3β/Snail pathway [15][16]. Endosomal-associated protein RAB5 promotes tumor cell migration and invasion, focal adhesion turnover, and integrin trafficking of cancer cells [17][18]. RAB21 controls integrin-mediated cell adhesion and motility of cervical cancer cells. Earlier studies paved a way to identify autophagy signaling proteins as a target to map their interaction with endocytic proteins and cross-regulation in tumor progression. However, identifying such targets is still challenging. Targeting the most viable endocytosis-associated gene(s) may help to achieve this goal. One of the recent studies used RNAseq expression in Rubcn knockout (KO) and wildtype (WT) group (GSE118019) [19]. RWesearchers also analyzed Rubcn gene expression in two different conditions to further explore the list of gene signatures, pathways enriched in the presence as well as absence of Rubcn. In order to understand the phagocytosis in tumors and their consequences, rwesearhcers performed gene set enrichment analysis (GSEA) (Figure 1). Interestingly, in GSEA, rwesearchers identified several important pathways such as apoptosis, glycolysis, hypoxia, and IFNγ response enriched in KO group (Figure 2). Similarly, using differential gene expression analysis, rwesearchers identified several important genes such as Rab4a, Gzme, Glrp1 that were upregulated in Rubcn KO group. This analysis suggests that the knock-out of endocytosis-associated genes also accelerates a more immunogenic microenvironment.
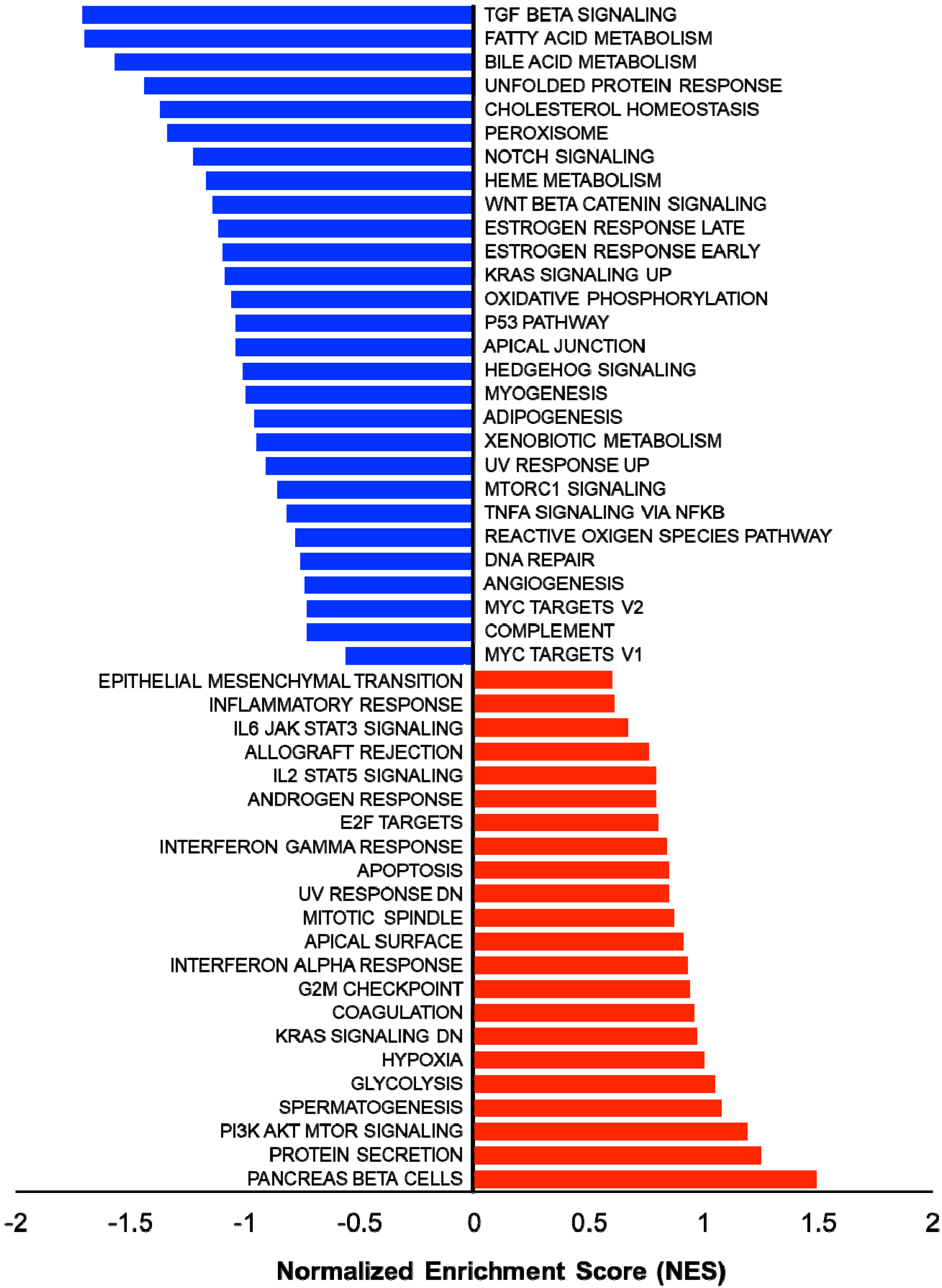
Figure 1. Gene set enrichment analysis (GSEA) showing enriched gene sets of Rubcn KO and WT group comparison. Bars in red (positive NES) indicate significant enrichment in KO phenotype and bars in blue indicate WT phenotype (negative NES). Data source available fromGSE118019 [2].
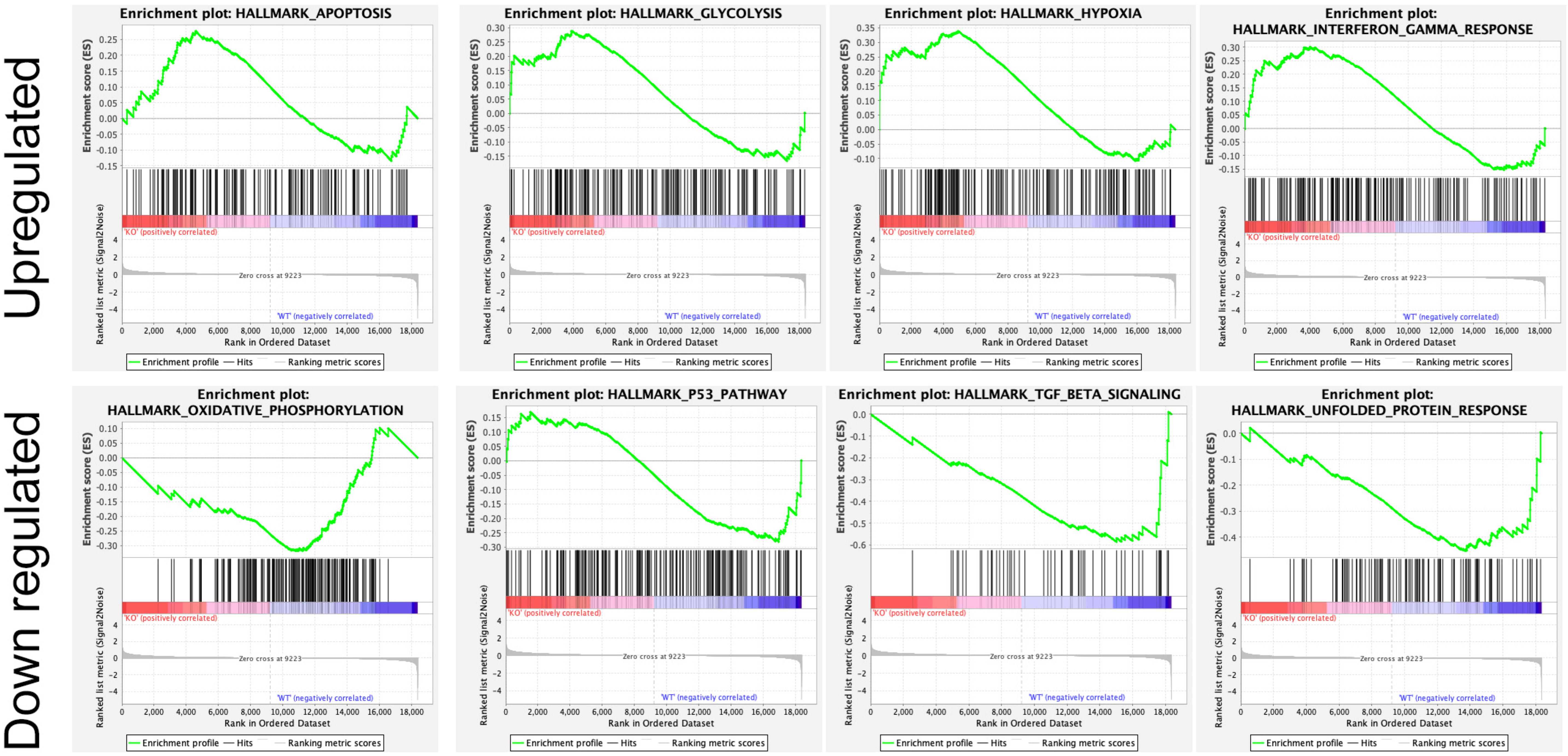
Figure 2. Enrichment plot of top four GSEA hallmarks enriched in Rubcn knockout and wildtype. Rubcn KO group enriched with significant upregulation in apoptosis, glycolysis, hypoxia, and interferon gamma response gene sets (upper panel). Oxidative phosphorylation, p53, TGFβ signaling, and unfolded protein response enriched in WT group (lower panel).
Cancer cells are hyper-proliferative and metastasize to the different part of the body. As a result of malignant proliferation, tumor cells need a rapid supply of nutrients for sustained proliferation. Macropinocytosis is a ligand-receptor-independent process and is exploited by cancer cells for rapid nutrient acquisition [20]. An earlier study showed that pancreatic cancer cell line KRPC was able to proliferate in the absence of essential amino acids in the culture media but obtain amino acids through macropinocytosis from extracellularly degraded albumin protein [21]. Macropinocytosis pathway can also facilitate the internalization of essential molecules, such as ATP, to mediate cancer cell proliferation and survival [22]. Macropinocytosis is also involved in K-Ras-mTORC1 signaling and may induce sustained mTORC1 activation and cell proliferation in cancer cells [23]. Moreover, macropinocytosis has been also reported in cell death of glioblastoma cancer cells with constitutive H-Ras activation in glioblastoma cell line U251, resulting in the accumulation macropinosomes and vacuolization of the cells [24].
2. Cardiovascular Disease
Cardiovascular diseases such as hypertension, coronary heart disease, stroke, and heart failure are the leading causes of mortality and morbidity [25]. Several endocytic proteins including sorting nexin (SNX), epsins, and disabled homolog 2 (Dab2) play an indispensable role in cardiovascular diseases [26]. SNX is a group of cytoplasmic and membrane-associated phosphoinositide binding proteins that play a role in protein trafficking [27][28]. Impairment of SNX pathway is responsible for the development of various forms of cardiovascular disease (CVD) [26]. In addition, SNX gene variants are also linked to CVD. Accumulating reports revealed that SNX exhibits its function by regulating expression and function of G protein-coupled receptors (GPCRs) such as receptor tyrosine kinases (RTKs) and dopamine receptors for the maintenance of blood pressure [29][30]. A previous report demonstrates that an impairment in the structure and function of SNX is associated with hypertension. Renal SNX5 expression regulates insulin degradation enzyme (IDE) activity and is associated with blood insulin and glucose levels [31]. Decreased renal expression of SNX5 expression leads to further elevation of systolic blood pressure and inhibition of sodium excretion [29]. Further, studies have also established the association of other SNX, such as SNX19, with coronary artery disease. However, the mechanistic pathways behind the SNXs-induced coronary artery disease remain enigmatic [26]. Moreover, SNXs may play a key role in coronary artery pathogenesis by regulating lipid metabolism. The influence of SNXs on a lipid level may be because of the interaction of SNX1, SNX2, and SNX4 with leptin receptors [32]. Furthermore, studies also suggested the abnormal expression of SNX leads to heart failure. Endogenous SNX13 level reduced in failing the heart of mice and human. In SNX13-deficient zebra fish, decreased cardiac systolic function was associated with cardiomyocyte apoptosis, and an inhibition of which improves the cardiac dysfunction [33]. In cardiovascular disease, atherosclerosis is a crucial player associated to morbidity and mortality. Epsins are endocytic adaptor proteins associated with cardiovascular disease [34]. Epsins are involved in endothelial cell dysfunction as well initiation and progression of arthrosclerosis through interaction with inositol 1,4,5 triphosphate receptor type 1 (IP3R1) [34]. Furthermore, Dab2, a multifactorial protein, plays a vital role in several cellular functions, including cell adhesion, cell signaling, and endocytosis. More importantly, Dab2 is associated with cholesterol metabolism and low-density lipoprotein (LDL) uptake by regulating the LDL receptor endocytosis. An earlier study suggests that deletion of Dab2 in liver endothelial cells results in an elevated level of serum LDL and cholesterol [35]. An earlier study shown that the Dab2 gene variant is associated with increased risk of coronary artery disease [36]. Interestingly, another report showed that quercetin-mediated up-regulation of Dab2 expression attenuates the atherosclerosis [37]. Hence, Dab2 is considered as a new anti-atherosclerosis therapeutic.
Earlier reports suggested that the CD36 (cluster of differentiation 36), a transmembrane glycoprotein receptor, plays an important role in athero-thrombotic activity and promotes the pathological conditions such as atherosclerosis and thrombosis [38]. CD36 is a pattern recognition receptor (PRR) and multi-functional protein that is majorly involved in the uptake of fatty acids (FA) in adipose tissues and plays a key role in regulation of lipid metabolism [39]. The process of FA uptake and its delivery is facilitated by caveolae-dependent internalization of CD36. Additionally, the FA uptake mediated by CD36 is a palmitoylation-regulated endocytic pathway [40]. The CD36 is expressed on the surface of various cell types including skeletal and cardiac myocytes [41]. It acts as a key player in energetics of cardiac myocytes as it facilitates FA transport, which further utilizes beta oxidation and leads to energy generation. Apart from FA, CD36 also recognizes and interacts with oxidized LDL (oxLDL), which eventually progresses atherosclerosis [42]. The oxLDL has been shown to promote apoptosis signaling in the vascular smooth muscle cells and contributes to atherosclerotic plaques [43]. The expression of CD36 on macrophages and platelets also promotes signaling cascades of inflammation which eventually participates in atherosclerotic arterial lesion formation and thrombus formation [44]. Collectively, CD36 dysfunction has been shown to contribute to the pathologies of atherosclerosis. Therefore, targeting the CD36-mediated transport of lipid moieties could be an effective therapeutic approach for the treatment of atherosclerosis and thrombosis.
Moreover, the intracellular compartmentalization of G protein-coupled receptor (GPCR) during early endosome and Golgi apparatus distribution are associated with cardiovascular outcomes. Endosomal G protein signaling by vasopressin type 2 receptor (V2R) plays a key role in cardiac arrest. There are three types of vasopressin receptors, including V1AR, V1BR, and V2R, which are triggered by arginine vasopressin (AVP). An elevated level of AVP plays a crucial role in changing the cardiovascular function and impaired renal solute-free water excretion result in hyponatremia [45][46]. Therefore, vasopressin receptors, V2R, have emerged as a popular target to develop the antagonist against therapeutics for cardiac arrest and hyponatremia.
3. Neurological Disorders
Millions of neurons organize and perform the regular functioning of the brain and nervous system. The fundamental role of neurons includes the transmission and receiving of the information or signals and behaving as a unified structure. This communication of information among the neurons is possible due to the presence of junction-like structures, known as synapses. The communication among neurons occurs by electrical or chemical signals. The electrical signal relies on the phenomena known as action potential. The chemical signals are generated through the transmission of various chemicals among neurons. Broadly, these chemicals are defined as neurotransmitters. The neurotransmitters are stored inside the vesicle structures and tend to release at the synaptic cleft of the synapse for the transmission of the information [47]. At the synapse, the release of neurotransmitters relies on the two fundamental biological events, such as exocytosis and endocytosis. The process of exocytosis is responsible for the release of neurotransmitters, while the process of endocytosis is responsible for the recycling of the synaptic vesicle membranes [48].
Several events occur during the process of chemical neurotransmission, such as formation of synaptic vesicles (SV), fusion with plasma membrane for releasing neurotransmitters, and recycling of synaptic vesicles. The recycling of synaptic vesicles is the hallmark event which comprises three key steps at the synapse; (i) release of neurotransmitters, (ii) clathrin-mediated endocytosis, and (iii) ultrafast endocytosis [49]. Apart from those mechanisms, several other mechanisms, such as ultrafast bulk endocytosis and activity-dependent bulk endocytosis, are also responsible for the recycling of the vesicles [50][51]. Numerous proteins play vital roles in the mechanisms of endocytosis, including amphiphysin 1 (AMPH1), endophilin A1, clathrin, dynamin, and synaptojanin 1 (SYNJ1) [49]. Moreover, several regulatory proteins are also involved in the regulation of endocytosis. Collectively, these proteins function in an organized manner to propel various endocytic mechanisms, thus efficiently recycling the SVs, which is crucial for continuous supply of neurotransmitter-filled SVs, performance of sensory functions and maintenance of synaptic physiology [50]. These reports suggest that the process of endocytosis and associated proteins plays a significant role in neurotransmission at synapse and maintain the neural homeostasis.
Alterations in neuronal homeostasis and poor neuronal function lead to several pathological conditions collectively known as neurodegenerative diseases. In the current scenario, neurodegenerative diseases occur prominently in a large population of the world and pose a socio-economic burden [52]. Extensive studies suggest that the dysfunction of endocytosis signaling at synapse participates in the progression of various neurological disorders such as Alzheimer’s disease (AD), Parkinson’s disease, and Amyotrophic lateral sclerosis (ALS).
Alzheimer’s disease (AD) is the most prevalent type of neurodegenerative disease [53]. The major pathologies associated with AD include accumulation of amyloid-β (Aβ) plaques and development of neurofibrillary tangles (NFTs) due to hyper-phosphorylated tau protein [54]. Deregulation of endocytic processes such as CME and CIE accumulates the (Aβ) plaques and progresses AD [55]. The genome-wide association studies (GWAS) of AD patients revealed that the deregulated expression of genes PICALM, BIN-1, and sorLA, which are essentially involved in clathrin-mediated endocytosis [56][57]. In addition, the altered expression of Rab5, clathrin, dynamin 2, and PICALM has been found in transgenic Tg2576 mice AD experimental models [53][58]. Further, an elevated expression of caveolin-1 reported in the hippocampus and cortex regions of AD brains [59]. The endocytic signaling is considered as tightly regulated cellular process and vulnerable to hyperphosphorylated tau protein [60]. Apart from endocytic proteins and signaling, Endolysosomal or autophagic abnormalities play essential roles in the progression of AD pathologies [60]. The endocytic signaling is also involved in the internalization of extracellular Aβ that accumulates in endosomes and leads to neuronal toxicity [61]. Earlier evidence suggests that the amyloid precursor protein (APP) (a major component involved in the production of Aβ) could be internalized via CME and CIE pathways. Further, the expression level of endocytic proteins, APP, Tau proteins, and other molecules is varied with the age as well as AD progression. The expression of endocytic proteins such as AP180, caveolin-2, clathrin, dynamin-1, flotillin-2, and Rab-5 has been found significantly elevated, and that change accelerates endocytosis and progresses AD in aged brains [53]. Moreover, elevated tau protein induces microtubule assembly and sequesters free dynamins that impair the endocytosis and subsequently perturb the neurotransmission. The neural cells express N-methyl-D-aspartate (NMDA) receptors which are ionotropic glutamate receptors and regulate transmission of glutamate neurotransmitters. The surface expression of NMDA receptors is tightly regulated through clathrin-dependent endocytosis [62][63] and is shown to be endocytosed both in primary neuronal cultures and in vitro heterologous cells [62]. NMDA receptors play an important role in Aβ-induced neurotoxicity [62]. Moreover, Aβ regulates NMDA receptor response by promoting their endocytosis and are associated with synaptic transmission [64]. Therefore, the evidence suggests that the alteration in the endocytosis process and associated protein expression progresses AD.
Parkinson’s disease (PD) is the second most prevalent neurodegenerative disease associated with degeneration and subsequent loss of dopaminergic neurons [65][66]. The synaptic dysfunction is a key event prior to the loss of dopaminergic neurons and participates in the pathogenesis of PD. In a normal human brain, the neurotransmitters release as well as uptake and synaptic vesicles (SV) recycling occur in the synapse. The clathrin-mediated endocytosis participates in SV recycling [51]. Further, the process of synaptic vesicle endocytosis (SVE) regenerates a synaptic vesicle, which is a tightly regulated event and essential for neurotransmission. During PD, the synaptic dysfunction is associated with deregulated SVE signaling. Several genetic studies and mutation analysis suggest that the genes such as DNAJC6, SYNJ1, SH3GL2, SNCA, LRRK2, PRKN, and DJ-1 plays a vital role in the modulation of SVE and progression of PD [49]. The SVE dysfunction leads to erroneous dopamine packaging into the vesicles, as a consequence of elevated cytosolic dopamine and subsequent dopaminergic neurodegeneration [49]. Thus, deregulated expression of endocytic genes and SVE signaling perturb dopamine signaling and subsequent neurotransmission, which promotes the pathologies of PD. α-Synuclein (α-syn) is a 140-amino-acid soluble acidic protein highly expressed in pre-synaptic nerves has been implicated in the pathogenesis of PD [67]. α-syn can regulate clathrin-mediated endocytosis of membrane receptors [68] and is involved in the regulation of NMDA receptor endocytosis [69]. Further studies are needed to explore the mechanistic and therapeutic potential of targeting α-syn and NMDA receptors for the treatment of PD.
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease associated with the central nervous system (CNS). The major pathological condition of ALS is the degeneration of the motor neurons in CNS leading to muscles weakness [70]. Occurrence of ALS is sporadic and familial event, and multiple genes are involved in the progression of both types of ALS [71]. The genetic studies revealed that the mutation in the Chromosome 9 open reading frame 72 (C9ORF72) progresses ALS. Interestingly, the impaired endocytic signaling has been observed during C9ORF72 mutated conditions in C9ORF72 ALS/FTD patients as well as the SH-SY5Y cell line model, suggesting that the expression of C9ORF72 modulates endocytosis [72][73]. Further, the expression of C9ORF72 is associated with endocytosis of tropomyosin receptor kinase receptor B (TrkB) (essential for the development and functioning of nervous system) in neurons [70]. Earlier reports suggested that the valosin-containing protein (VCP/p97) regulates endolysosomal sorting of ubiquitylated caveolin-1 [74]. Another protein TDP-43 (nuclear RNA binding protein) inhibits endocytosis and localizes with endocytic proteins in tissue samples of ALS patients. In addition, dynamics of endocytosis modulate TDP-43 expression in a TDP-43 ALS fly model [75].
Collectively, these studies suggest that various endocytic pathways play an essential role in the progression of neurodegenerative diseases. Therefore, targeting the endocytic proteins and signaling mechanism associated with endocytic processes could be the effective approach to target neurological diseases for therapeutic intervention.
4. Inflammatory Bowel Diseases
Epithelial integrity and barrier function are critical to separate luminal contents, including nutrients and microbes from the underlying intestinal tissues [76][77]. Perturbations to the epithelial integrity are believed to contribute intestinal dysbiosis and allows heightened microbial penetration, resulting in chronic diseases such as inflammatory bowel disease (IBD), which is an umbrella term for Crohn’s disease (CD) and ulcerative colitis (UC) [78][79]. Endocytotic exodus of microbes in the barrier dysfunction CD, induces mucosal inflammation [80][81]. A case study of a 24-year-old woman with a positive family history of Crohn’s disease showed an increased intestinal permeability precedes the onset of Crohn’s disease [82]. Accumulating evidence shows that the intestinal barrier integrity is chiefly regulated by several multicomplex proteins, constituted of tight junctions (TJs) and adherens junctions (AJs) proteins [83]. Mammalian TJs have diverse roles, ranging from mediating selective diffusion of molecules across the epithelium to cis and trans interactions at sites of intercellular spaces [84]. The claudin protein family of TJs, including junctional adhesion molecule (JAM)-A, the tight junction-associated MARVEL proteins (TAMP), and coxsackievirus and adenovirus receptor (CAR), additionally consist of scaffolding molecules such as the zonula occludens (ZO) protein [85], chiefly regulates the organizational framework of intercellular barrier [86]. These claudin protein family are fundamental to establishing the paracellular passages of nutrients between the intestinal lumen and internal environment, as well as defense mechanisms against pathogens [87][88]. Dysregulated expression of sealing claudins and increased intestinal permeability contributes to a leaky epithelial barrier and may lead to intestinal infection and bowel symptoms of IBD patients [78][89]. The dextran sulfate sodium (DSS) colitis has been described as a most suitable in vivo experimental model of IBD to study intestinal barrier permeability and dissemination of microbes across the intestinal lumen [90][91][92]. Previously in the mouse DSS colitis model, a redistribution of occludin expression was observed compared to distinct appearance at the tight junctions of the apical membrane of colonic epithelium [93]. Apart from claudins, expression levels of other TJ proteins, such as JAM-A, occludin, and ZO-1, remain suppressed during intestinal inflammation [94].
Adherens junctions (AJs) are cell–cell adhesion complexes and usually annotated as “cadherins” [95]. Intestinal epithelial cells (IECs) largely express epithelial (E) cadherin, which is required to maintain colonic epithelial barrier permeability, and dysfunction can aggravate colitis [96]. Moreover, dysregulated expression of E-cadherin has been reported in tissue biopsies of IBD patients [97].
The proper assembly and functioning of junctional proteins are channelized through exocytic transportation of newly synthesized proteins to the cell surface and recycling of mature TJs and AJs via endocytosis [98]. The defects in TJs and AJs endocytosis leading to barrier disruption in IBD have been reported long ago [99]. The endocytosis of TJs and AJs happens similar to the internalization of luminal antigens into the enterocytes, and IBD patients have an increased ability to transcytosis of luminal antigens [89]. Importantly, recycling and exocytic trafficking of TJs and AJs proteins orchestrated through interaction of carrier vesicles and intracellular organelles [98]. Mechanistically, the membrane-targeted trafficking of carrier vesicles mediated through the protein family of Rab small GTPases and ultimate engrafting to lipid membrane is driven by the SNARE (soluble N-ethylmaleimide-sensitive factor associated receptor) proteins [100]. These results highlight an involvement of organelle-specific trafficking in the establishment of the intestinal epithelial barrier. The intracellular network of endoplasmic reticulum (ER)-Golgi trafficking controls synthesis and recycling of junctional proteins during intestinal inflammation [101]. Notably, the excessive fragmentation and vascularization are the characteristic dysfunction of the ER and the Golgi networks and observed in the intestinal mucosa of UC patients [102].
Altogether, an intact mucosal barrier restricts the infiltration of pathogenic microbes and regulates the absorption and passage of nutrients from the intestinal lumen into the underlying circulation. Endocytosis plays a critical role in the trafficking of intestinal junctional proteins TJs and AJs; however, dysfunction of the junctional proteins is a causative factor in the pathogenesis of IBD. Additional studies may further elucidate the mol
References
- Lanzetti, L.; Di Fiore, P.P. Endocytosis and cancer: An ‘insider’ network with dangerous liaisons. Traffic 2008, 9, 2011–2021.
- Rajendran, B.K.; Deng, C.X. Characterization of potential driver mutations involved in human breast cancer by computational approaches. Oncotarget 2017, 8, 50252–50272.
- Rajendran, B.K.; Deng, C.X. A comprehensive genomic meta-analysis identifies confirmatory role of OBSCN gene in breast tumorigenesis. Oncotarget 2017, 8, 102263–102276.
- Colaluca, I.N.; Tosoni, D.; Nuciforo, P.; Senic-Matuglia, F.; Galimberti, V.; Viale, G.; Pece, S.; Di Fiore, P.P. NUMB controls p53 tumour suppressor activity. Nature 2008, 451, 76–80.
- Ding, R.B.; Chen, P.; Rajendran, B.K.; Lyu, X.; Wang, H.; Bao, J.; Zeng, J.; Hao, W.; Sun, H.; Wong, A.H.; et al. Molecular landscape and subtype-specific therapeutic response of nasopharyngeal carcinoma revealed by integrative pharmacogenomics. Nat. Commun. 2021, 12, 3046.
- Khan, I.; Steeg, P.S. Endocytosis: A pivotal pathway for regulating metastasis. Br. J. Cancer 2021, 124, 66–75.
- Beck, B.H.; Welch, D.R. The KISS1 metastasis suppressor: A good night kiss for disseminated cancer cells. Eur. J. Cancer 2010, 46, 1283–1289.
- Dawson, J.C.; Timpson, P.; Kalna, G.; Machesky, L.M. Mtss1 regulates epidermal growth factor signaling in head and neck squamous carcinoma cells. Oncogene 2012, 31, 1781–1793.
- Engelman, J.A.; Zhang, X.L.; Galbiati, F.; Lisanti, M.P. Chromosomal localization, genomic organization, and developmental expression of the murine caveolin gene family (Cav-1, -2, and -3). Cav-1 and Cav-2 genes map to a known tumor suppressor locus (6-A2/7q31). FEBS Lett. 1998, 429, 330–336.
- Joshi, B.; Strugnell, S.S.; Goetz, J.G.; Kojic, L.D.; Cox, M.E.; Griffith, O.L.; Chan, S.K.; Jones, S.J.; Leung, S.P.; Masoudi, H.; et al. Phosphorylated caveolin-1 regulates Rho/ROCK-dependent focal adhesion dynamics and tumor cell migration and invasion. Cancer Res. 2008, 68, 8210–8220.
- Azarnia Tehran, D.; Lopez-Hernandez, T.; Maritzen, T. Endocytic Adaptor Proteins in Health and Disease: Lessons from Model Organisms and Human Mutations. Cells 2019, 8, 1345.
- Boulay, P.L.; Schlienger, S.; Lewis-Saravalli, S.; Vitale, N.; Ferbeyre, G.; Claing, A. ARF1 controls proliferation of breast cancer cells by regulating the retinoblastoma protein. Oncogene 2011, 30, 3846–3861.
- Schlienger, S.; Ramirez, R.A.; Claing, A. ARF1 regulates adhesion of MDA-MB-231 invasive breast cancer cells through formation of focal adhesions. Cell Signal. 2015, 27, 403–415.
- Schlienger, S.; Campbell, S.; Pasquin, S.; Gaboury, L.; Claing, A. ADP-ribosylation factor 1 expression regulates epithelial-mesenchymal transition and predicts poor clinical outcome in triple-negative breast cancer. Oncotarget 2016, 7, 15811–15827.
- Chang, Y.C.; Su, C.Y.; Chen, M.H.; Chen, W.S.; Chen, C.L.; Hsiao, M. Secretory RAB GTPase 3C modulates IL6-STAT3 pathway to promote colon cancer metastasis and is associated with poor prognosis. Mol. Cancer 2017, 16, 135.
- Yang, J.; Liu, W.; Lu, X.; Fu, Y.; Li, L.; Luo, Y. High expression of small GTPase Rab3D promotes cancer progression and metastasis. Oncotarget 2015, 6, 11125–11138.
- Mendoza, P.; Ortiz, R.; Diaz, J.; Quest, A.F.; Leyton, L.; Stupack, D.; Torres, V.A. Rab5 activation promotes focal adhesion disassembly, migration and invasiveness in tumor cells. J. Cell Sci. 2013, 126, 3835–3847.
- Diaz, J.; Mendoza, P.; Ortiz, R.; Diaz, N.; Leyton, L.; Stupack, D.; Quest, A.F.; Torres, V.A. Rab5 is required in metastatic cancer cells for Caveolin-1-enhanced Rac1 activation, migration and invasion. J. Cell Sci. 2014, 127, 2401–2406.
- Cunha, L.D.; Yang, M.; Carter, R.; Guy, C.; Harris, L.; Crawford, J.C.; Quarato, G.; Boada-Romero, E.; Kalkavan, H.; Johnson, M.D.L.; et al. LC3-Associated Phagocytosis in Myeloid Cells Promotes Tumor Immune Tolerance. Cell 2018, 175, 429–441e.16.
- Zhang, Y.; Commisso, C. Macropinocytosis in Cancer: A Complex Signaling Network. Trends Cancer 2019, 5, 332–334.
- Kamphorst, J.J.; Nofal, M.; Commisso, C.; Hackett, S.R.; Lu, W.; Grabocka, E.; Vander Heiden, M.G.; Miller, G.; Drebin, J.A.; Bar-Sagi, D.; et al. Human pancreatic cancer tumors are nutrient poor and tumor cells actively scavenge extracellular protein. Cancer Res. 2015, 75, 544–553.
- Recouvreux, M.V.; Commisso, C. Macropinocytosis: A Metabolic Adaptation to Nutrient Stress in Cancer. Front. Endocrinol. 2017, 8, 261.
- Yoshida, S.; Pacitto, R.; Inoki, K.; Swanson, J. Macropinocytosis, mTORC1 and cellular growth control. Cell. Mol. Life Sci. 2018, 75, 1227–1239.
- Overmeyer, J.H.; Kaul, A.; Johnson, E.E.; Maltese, W.A. Active ras triggers death in glioblastoma cells through hyperstimulation of macropinocytosis. Mol. Cancer Res. 2008, 6, 965–977.
- Benjamin, E.J.; Virani, S.S.; Callaway, C.W.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Chiuve, S.E.; Cushman, M.; Delling, F.N.; Deo, R.; et al. Heart Disease and Stroke Statistics-2018 Update: A Report From the American Heart Association. Circulation 2018, 137, e67–e492.
- Yang, J.; Villar, V.A.M.; Rozyyev, S.; Jose, P.A.; Zeng, C. The emerging role of sorting nexins in cardiovascular diseases. Clin. Sci. 2019, 133, 723–737.
- Teasdale, R.D.; Collins, B.M. Insights into the PX (phox-homology) domain and SNX (sorting nexin) protein families: Structures, functions and roles in disease. Biochem. J. 2012, 441, 39–59.
- Chandra, M.; Collins, B.M. The Phox Homology (PX) Domain. Adv. Exp. Med. Biol. 2019, 1111, 1–17.
- Villar, V.A.; Armando, I.; Sanada, H.; Frazer, L.C.; Russo, C.M.; Notario, P.M.; Lee, H.; Comisky, L.; Russell, H.A.; Yang, Y.; et al. Novel role of sorting nexin 5 in renal D(1) dopamine receptor trafficking and function: Implications for hypertension. FASEB J. 2013, 27, 1808–1819.
- Villar, V.A.; Jones, J.E.; Armando, I.; Asico, L.D.; Escano, C.S., Jr.; Lee, H.; Wang, X.; Yang, Y.; Pascua-Crusan, A.M.; Palmes-Saloma, C.P.; et al. Sorting nexin 1 loss results in D5 dopamine receptor dysfunction in human renal proximal tubule cells and hypertension in mice. J. Biol. Chem. 2013, 288, 152–163.
- Li, F.; Yang, J.; Villar, V.A.M.; Asico, L.D.; Ma, X.; Armando, I.; Sanada, H.; Yoneda, M.; Felder, R.A.; Jose, P.A.; et al. Loss of renal SNX5 results in impaired IDE activity and insulin resistance in mice. Diabetologia 2018, 61, 727–737.
- Haft, C.R.; de la Luz Sierra, M.; Barr, V.A.; Haft, D.H.; Taylor, S.I. Identification of a family of sorting nexin molecules and characterization of their association with receptors. Mol. Cell. Biol. 1998, 18, 7278–7287.
- Li, J.; Li, C.; Zhang, D.; Shi, D.; Qi, M.; Feng, J.; Yuan, T.; Xu, X.; Liang, D.; Xu, L.; et al. SNX13 reduction mediates heart failure through degradative sorting of apoptosis repressor with caspase recruitment domain. Nat. Commun. 2014, 5, 5177.
- Cui, K.; Dong, Y.; Wang, B.; Cowan, D.B.; Chan, S.L.; Shyy, J.; Chen, H. Endocytic Adaptors in Cardiovascular Disease. Front. Cell Dev. Biol. 2020, 8, 624159.
- Tao, W.; Moore, R.; Smith, E.R.; Xu, X.X. Endocytosis and Physiology: Insights from Disabled-2 Deficient Mice. Front. Cell Dev. Biol. 2016, 4, 129.
- Wang, Y.; Wang, Y.; Adi, D.; He, X.; Liu, F.; Abudesimu, A.; Fu, Z.; Ma, Y. Dab2 gene variant is associated with increased coronary artery disease risk in Chinese Han population. Medicine 2020, 99, e20924.
- Lin, W.; Wang, W.; Wang, D.; Ling, W. Quercetin protects against atherosclerosis by inhibiting dendritic cell activation. Mol. Nutr. Food Res. 2017, 61, 1700031.
- Silverstein, R.L.; Li, W.; Park, Y.M.; Rahaman, S.O. Mechanisms of cell signaling by the scavenger receptor CD36: Implications in atherosclerosis and thrombosis. Trans. Am. Clin. Climatol. Assoc. 2010, 121, 206–220.
- Coburn, C.T.; Knapp, F.F., Jr.; Febbraio, M.; Beets, A.L.; Silverstein, R.L.; Abumrad, N.A. Defective uptake and utilization of long chain fatty acids in muscle and adipose tissues of CD36 knockout mice. J. Biol. Chem. 2000, 275, 32523–32529.
- Hao, J.W.; Wang, J.; Guo, H.; Zhao, Y.Y.; Sun, H.H.; Li, Y.F.; Lai, X.Y.; Zhao, N.; Wang, X.; Xie, C.; et al. CD36 facilitates fatty acid uptake by dynamic palmitoylation-regulated endocytosis. Nat. Commun. 2020, 11, 4765.
- Febbraio, M.; Hajjar, D.P.; Silverstein, R.L. CD36: A class B scavenger receptor involved in angiogenesis, atherosclerosis, inflammation, and lipid metabolism. J. Clin. Invest. 2001, 108, 785–791.
- Matsuura, E.; Hughes, G.R.; Khamashta, M.A. Oxidation of LDL and its clinical implication. Autoimmun. Rev. 2008, 7, 558–566.
- Okura, Y.; Brink, M.; Itabe, H.; Scheidegger, K.J.; Kalangos, A.; Delafontaine, P. Oxidized low-density lipoprotein is associated with apoptosis of vascular smooth muscle cells in human atherosclerotic plaques. Circulation 2000, 102, 2680–2686.
- Park, Y.M. CD36, a scavenger receptor implicated in atherosclerosis. Exp. Mol. Med. 2014, 46, e99.
- Goldsmith, S.R. Arginine vasopressin antagonism in heart failure: Current status and possible new directions. J. Cardiol. 2019, 74, 49–52.
- Finley, J.J.t.; Konstam, M.A.; Udelson, J.E. Arginine vasopressin antagonists for the treatment of heart failure and hyponatremia. Circulation 2008, 118, 410–421.
- Gross, O.P.; von Gersdorff, H. Recycling at synapses. Elife 2016, 5, e17692.
- Saheki, Y.; De Camilli, P. Synaptic vesicle endocytosis. Cold Spring Harb. Perspect. Biol. 2012, 4, a005645.
- Zou, L.; Tian, Y.; Zhang, Z. Dysfunction of Synaptic Vesicle Endocytosis in Parkinson’s Disease. Front. Integr. Neurosci. 2021, 15, 619160.
- Milosevic, I. Revisiting the Role of Clathrin-Mediated Endoytosis in Synaptic Vesicle Recycling. Front. Cell. Neurosci. 2018, 12, 27.
- Chanaday, N.L.; Cousin, M.A.; Milosevic, I.; Watanabe, S.; Morgan, J.R. The Synaptic Vesicle Cycle Revisited: New Insights into the Modes and Mechanisms. J. Neurosci. 2019, 39, 8209–8216.
- Zahra, W.; Rai, S.N.; Birla, H.; Singh, S.S.; Dilnashin, H.; Rathore, A.S.; Singh, S.P. The Global Economic Impact of Neurodegenerative Diseases: Opportunities and Challenges. In Bioeconomy for Sustainable Development; Keswani, C., Ed.; Springer: Singapore, 2020; pp. 333–345.
- Alsaqati, M.; Thomas, R.S.; Kidd, E.J. Proteins Involved in Endocytosis Are Upregulated by Ageing in the Normal Human Brain: Implications for the Development of Alzheimer’s Disease. J. Gerontol. Ser. A 2018, 73, 289–298.
- Citron, M. Strategies for disease modification in Alzheimer’s disease. Nat. Rev. Neurosci. 2004, 5, 677–685.
- Tate, B.A.; Mathews, P.M. Targeting the role of the endosome in the pathophysiology of Alzheimer’s disease: A strategy for treatment. Sci. Aging Knowl. Environ. 2006, 2006, re2.
- McMahon, H.T.; Boucrot, E. Molecular mechanism and physiological functions of clathrin-mediated endocytosis. Nat. Rev. Mol. Cell Biol. 2011, 12, 517–533.
- Lambert, J.C.; Ibrahim-Verbaas, C.A.; Harold, D.; Naj, A.C.; Sims, R.; Bellenguez, C.; DeStafano, A.L.; Bis, J.C.; Beecham, G.W.; Grenier-Boley, B.; et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 2013, 45, 1452–1458.
- Thomas, R.S.; Lelos, M.J.; Good, M.A.; Kidd, E.J. Clathrin-mediated endocytic proteins are upregulated in the cortex of the Tg2576 mouse model of Alzheimer’s disease-like amyloid pathology. Biochem. Biophys. Res. Commun. 2011, 415, 656–661.
- Gaudreault, S.B.; Dea, D.; Poirier, J. Increased caveolin-1 expression in Alzheimer’s disease brain. Neurobiol. Aging 2004, 25, 753–759.
- Ando, K.; Houben, S.; Homa, M.; de Fisenne, M.A.; Potier, M.C.; Erneux, C.; Brion, J.P.; Leroy, K. Alzheimer’s Disease: Tau Pathology and Dysfunction of Endocytosis. Front. Mol. Neurosci. 2020, 13, 583755.
- Yang, A.J.; Chandswangbhuvana, D.; Margol, L.; Glabe, C.G. Loss of endosomal/lysosomal membrane impermeability is an early event in amyloid Abeta1-42 pathogenesis. J. Neurosci. Res. 1998, 52, 691–698.
- Roche, K.W.; Standley, S.; McCallum, J.; Dune Ly, C.; Ehlers, M.D.; Wenthold, R.J. Molecular determinants of NMDA receptor internalization. Nat. Neurosci. 2001, 4, 794–802.
- Petralia, R.S.; Wang, Y.X.; Wenthold, R.J. Internalization at glutamatergic synapses during development. Eur. J. Neurosci. 2003, 18, 3207–3217.
- Wang, Z.C.; Zhao, J.; Li, S. Dysregulation of synaptic and extrasynaptic N-methyl-D-aspartate receptors induced by amyloid-beta. Neurosci. Bull. 2013, 29, 752–760.
- de Lau, L.M.; Breteler, M.M. Epidemiology of Parkinson’s disease. Lancet Neurol. 2006, 5, 525–535.
- Kurowska, Z.; Kordower, J.H.; Stoessl, A.J.; Burke, R.E.; Brundin, P.; Yue, Z.; Brady, S.T.; Milbrandt, J.; Trapp, B.D.; Sherer, T.B.; et al. Is Axonal Degeneration a Key Early Event in Parkinson’s Disease? J. Park. Dis. 2016, 6, 703–707.
- Choudhury, S.P.; Bano, S.; Sen, S.; Suchal, K.; Kumar, S.; Nikolajeff, F.; Dey, S.K.; Sharma, V. Altered neural cell junctions and ion-channels leading to disrupted neuron communication in Parkinson’s disease. NPJ Park. Dis. 2022, 8, 66.
- Ben Gedalya, T.; Loeb, V.; Israeli, E.; Altschuler, Y.; Selkoe, D.J.; Sharon, R. Alpha-synuclein and polyunsaturated fatty acids promote clathrin-mediated endocytosis and synaptic vesicle recycling. Traffic 2009, 10, 218–234.
- Cheng, F.; Li, X.; Li, Y.; Wang, C.; Wang, T.; Liu, G.; Baskys, A.; Ueda, K.; Chan, P.; Yu, S. alpha-Synuclein promotes clathrin-mediated NMDA receptor endocytosis and attenuates NMDA-induced dopaminergic cell death. J. Neurochem. 2011, 119, 815–825.
- Burk, K.; Pasterkamp, R.J. Disrupted neuronal trafficking in amyotrophic lateral sclerosis. Acta Neuropathol. 2019, 137, 859–877.
- Otomo, A.; Pan, L.; Hadano, S. Dysregulation of the autophagy-endolysosomal system in amyotrophic lateral sclerosis and related motor neuron diseases. Neurol. Res. Int. 2012, 2012, 498428.
- Farg, M.A.; Sundaramoorthy, V.; Sultana, J.M.; Yang, S.; Atkinson, R.A.; Levina, V.; Halloran, M.A.; Gleeson, P.A.; Blair, I.P.; Soo, K.Y.; et al. C9ORF72, implicated in amytrophic lateral sclerosis and frontotemporal dementia, regulates endosomal trafficking. Hum. Mol. Genet. 2014, 23, 3579–3595.
- Burd, C.; Cullen, P.J. Retromer: A master conductor of endosome sorting. Cold Spring Harb. Perspect. Biol. 2014, 6, a016774.
- Ritz, D.; Vuk, M.; Kirchner, P.; Bug, M.; Schutz, S.; Hayer, A.; Bremer, S.; Lusk, C.; Baloh, R.H.; Lee, H.; et al. Endolysosomal sorting of ubiquitylated caveolin-1 is regulated by VCP and UBXD1 and impaired by VCP disease mutations. Nat. Cell Biol. 2011, 13, 1116–1123.
- Liu, G.; Coyne, A.N.; Pei, F.; Vaughan, S.; Chaung, M.; Zarnescu, D.C.; Buchan, J.R. Endocytosis regulates TDP-43 toxicity and turnover. Nat. Commun. 2017, 8, 2092.
- Yamada, T.; Sartor, R.B.; Marshall, S.; Specian, R.D.; Grisham, M.B. Mucosal injury and inflammation in a model of chronic granulomatous colitis in rats. Gastroenterology 1993, 104, 759–771.
- Mashimo, H.; Wu, D.C.; Podolsky, D.K.; Fishman, M.C. Impaired defense of intestinal mucosa in mice lacking intestinal trefoil factor. Science 1996, 274, 262–265.
- Chang, J.; Leong, R.W.; Wasinger, V.C.; Ip, M.; Yang, M.; Phan, T.G. Impaired Intestinal Permeability Contributes to Ongoing Bowel Symptoms in Patients With Inflammatory Bowel Disease and Mucosal Healing. Gastroenterology 2017, 153, 723–731e.21.
- Ranjan, K. Intestinal Immune Homeostasis and Inflammatory Bowel Disease: A Perspective on Intracellular Response Mechanisms. Gastrointest. Disord. 2020, 2, 24.
- Soderholm, J.D.; Streutker, C.; Yang, P.C.; Paterson, C.; Singh, P.K.; McKay, D.M.; Sherman, P.M.; Croitoru, K.; Perdue, M.H. Increased epithelial uptake of protein antigens in the ileum of Crohn’s disease mediated by tumour necrosis factor alpha. Gut 2004, 53, 1817–1824.
- Huang, C.; Hedl, M.; Ranjan, K.; Abraham, C. LACC1 Required for NOD2-Induced, ER Stress-Mediated Innate Immune Outcomes in Human Macrophages and LACC1 Risk Variants Modulate These Outcomes. Cell Rep. 2019, 29, 4525–4539e.24.
- Irvine, E.J.; Marshall, J.K. Increased intestinal permeability precedes the onset of Crohn’s disease in a subject with familial risk. Gastroenterology 2000, 119, 1740–1744.
- Giepmans, B.N.; van Ijzendoorn, S.C. Epithelial cell-cell junctions and plasma membrane domains. Biochim. Biophys. Acta 2009, 1788, 820–831.
- Gunzel, D.; Yu, A.S. Claudins and the modulation of tight junction permeability. Physiol. Rev. 2013, 93, 525–569.
- Ivanov, A.I.; Nusrat, A.; Parkos, C.A. Endocytosis of epithelial apical junctional proteins by a clathrin-mediated pathway into a unique storage compartment. Mol. Biol. Cell 2004, 15, 176–188.
- Furuse, M. Molecular basis of the core structure of tight junctions. Cold Spring Harb. Perspect. Biol. 2010, 2, a002907.
- Zhu, L.; Han, J.; Li, L.; Wang, Y.; Li, Y.; Zhang, S. Claudin Family Participates in the Pathogenesis of Inflammatory Bowel Diseases and Colitis-Associated Colorectal Cancer. Front. Immunol. 2019, 10, 1441.
- Mullin, J.M.; Marano, C.W.; Laughlin, K.V.; Nuciglio, M.; Stevenson, B.R.; Soler, P. Different size limitations for increased transepithelial paracellular solute flux across phorbol ester and tumor necrosis factor-treated epithelial cell sheets. J. Cell. Physiol. 1997, 171, 226–233.
- Landy, J.; Ronde, E.; English, N.; Clark, S.K.; Hart, A.L.; Knight, S.C.; Ciclitira, P.J.; Al-Hassi, H.O. Tight junctions in inflammatory bowel diseases and inflammatory bowel disease associated colorectal cancer. World J. Gastroenterol. 2016, 22, 3117–3126.
- Kang, J.W.; Yan, J.; Ranjan, K.; Zhang, X.; Turner, J.R.; Abraham, C. Myeloid Cell Expression of LACC1 Is Required for Bacterial Clearance and Control of Intestinal Inflammation. Gastroenterology 2020, 159, 1051–1067.
- Ranjan, K.; Hedl, M.; Abraham, C. The E3 ubiquitin ligase RNF186 and RNF186 risk variants regulate innate receptor-induced outcomes. Proc. Natl. Acad. Sci. USA 2021, 118, e2013500118.
- Ranjan, K.; Hedl, M.; Sinha, S.; Zhang, X.; Abraham, C. Ubiquitination of ATF6 by disease-associated RNF186 promotes the innate receptor-induced unfolded protein response. J. Clin. Investig. 2021, 131, e145472.
- Nighot, P.; Ma, T. Endocytosis of Intestinal Tight Junction Proteins: In Time and Space. Inflamm. Bowel Dis. 2021, 27, 283–290.
- Coeffier, M.; Gloro, R.; Boukhettala, N.; Aziz, M.; Lecleire, S.; Vandaele, N.; Antonietti, M.; Savoye, G.; Bole-Feysot, C.; Dechelotte, P.; et al. Increased proteasome-mediated degradation of occludin in irritable bowel syndrome. Am. J. Gastroenterol. 2010, 105, 1181–1188.
- Arrieta, M.C.; Bistritz, L.; Meddings, J.B. Alterations in intestinal permeability. Gut 2006, 55, 1512–1520.
- Grill, J.I.; Neumann, J.; Hiltwein, F.; Kolligs, F.T.; Schneider, M.R. Intestinal E-cadherin Deficiency Aggravates Dextran Sodium Sulfate-Induced Colitis. Dig. Dis. Sci. 2015, 60, 895–902.
- Kosovac, K.; Brenmoehl, J.; Holler, E.; Falk, W.; Schoelmerich, J.; Hausmann, M.; Rogler, G. Association of the NOD2 genotype with bacterial translocation via altered cell-cell contacts in Crohn’s disease patients. Inflamm. Bowel Dis. 2010, 16, 1311–1321.
- Fletcher, S.J.; Iqbal, M.; Jabbari, S.; Stekel, D.; Rappoport, J.Z. Analysis of occludin trafficking, demonstrating continuous endocytosis, degradation, recycling and biosynthetic secretory trafficking. PLoS ONE 2014, 9, e111176.
- Ivanov, A.I.; Nusrat, A.; Parkos, C.A. The epithelium in inflammatory bowel disease: Potential role of endocytosis of junctional proteins in barrier disruption. Novartis Found. Symp. 2004, 263, 115–124; discussion 124–132, 211–218.
- Burgoyne, R.D.; Morgan, A. Secretory granule exocytosis. Physiol. Rev. 2003, 83, 581–632.
- Yu, M.; Yang, S.; Qiu, Y.; Chen, G.; Wang, W.; Xu, C.; Cai, W.; Sun, L.; Xiao, W.; Yang, H. Par-3 modulates intestinal epithelial barrier function through regulating intracellular trafficking of occludin and myosin light chain phosphorylation. J. Gastroenterol. 2015, 50, 1103–1113.
- McGuckin, M.A.; Eri, R.; Simms, L.A.; Florin, T.H.; Radford-Smith, G. Intestinal barrier dysfunction in inflammatory bowel diseases. Inflamm. Bowel Dis. 2009, 15, 100–113.
More