Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Peter Tang and Version 1 by GU shiyan.
N6-metyladenosine (m6A), one of the most common RNA methylation modifications in mammals, has attracted extensive attentions owing to its regulatory roles in a variety of physiological and pathological processes. As a reversible epigenetic modification on RNAs, m6A is dynamically mediated by the functional interplay among the regulatory proteins of methyltransferases, demethylases and methyl-binding proteins. It has become increasingly clear that m6A modification is associated with the production and function of microRNAs (miRNAs).
- N6-metyladenosine
- methyltransferases
- demethylases
- methyl-binding proteins
- miRNAs
1. Introduction
N6-metyladenosine (m6A) modification refers to the methylation that occurs at the 6th nitrogen atom of adenine. Since firstly found in 1974, m6A has received extensive attention for regulating abundant internal modifications on various RNAs, including mRNAs, microRNAs (miRNAs), small nuclear RNAs (snRNAs), long non-coding RNAs (lncRNAs) and circular RNAs (circRNAs) [1,2,3][1][2][3]. As weis all known, the formation of m6A modification is dynamically catalyzed by methyltransferases and demethylases, also called “writers” and “erasers”, respectively. The binding proteins, named “readers”, can specifically combine with m6A modification and mediate m6A biological function in different pathological and physiological processes [4]. At present, m6A modification has been widely recognized as a reversible and dynamic epigenetic modification on various RNAs [5], and it is becoming increasingly clear that m6A potentially contributes to the occurrence and development of multiple diseases through altering RNA expressions or RNA functions [6]. Recently, the clinical value of m6A modification in diseases has become apparent, and m6A modification has been commonly utilized as a promising biomarker to diagnose, prevent and treat diseases [7].
As the most common non-coding RNAs, miRNAs exert their biological functions through interaction with RNAs or proteins [8]. Recently, emerging works of literature have demonstrated that miRNAs were modified by various chemical modifications, such as m6A, m1C, m5C and m7G, which affect the processing and functions of corresponding miRNAs [9,10,11][9][10][11]. Among these modifications, m6A attracted the most attention. As reported previous study, m6A modification is preferentially concentrated near the 3′-UTR, of which 67% contain ncRNAs, such as the binding sites of miRNAs, implicating that m6A and ncRNAs may jointly regulate target mRNAs through cooperation or competition [12]. The latest studies have found that m6A modification affects the cleavage, transport, stability and degradation of corresponding miRNAs and influences the interactions between miRNAs and long non-coding RNAs or proteins [13,14,15][13][14][15]. Interestingly, it is becoming increasingly clear that miRNAs also have critical roles in regulating m6A modification by changing the regulatory proteins of m6A modification [16].
2. Dynamic Regulation of m6A Modification
In recent years, more and more studies have found that m6A modification on RNAs can be dynamically regulated by dedicated methyltransferases, demethylases or methyl-binding proteins. A set of these regulatory proteins have been summarized in Figure 1.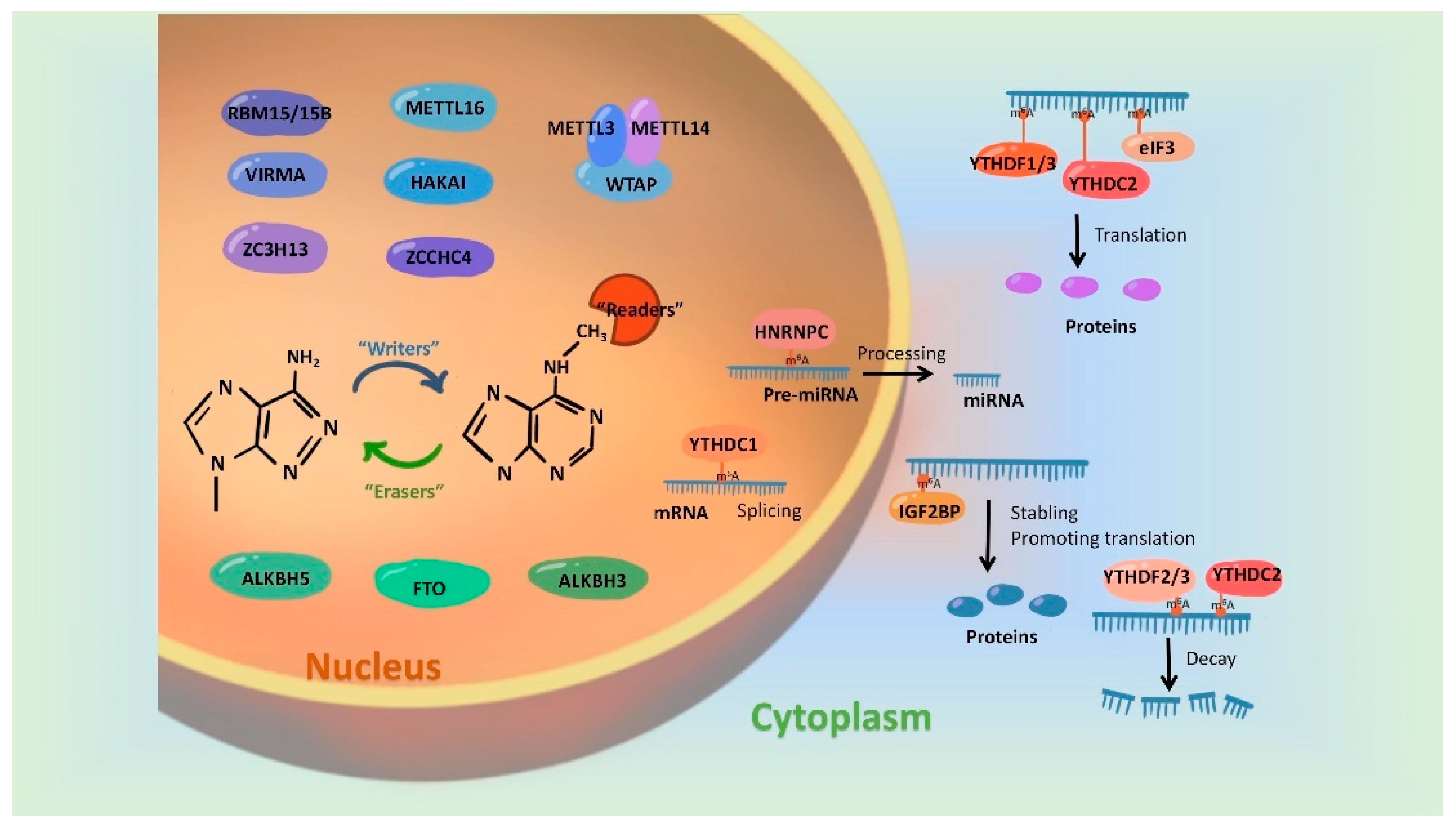
Figure 1. Regulatory proteins involved in mediating the m6A modification. m6A modification is synergistically catalyzed by methyltransferases (Writers), demethylases (Erasers) and methyl-binding proteins (Readers). The formation of m6A was initially catalyzed by a group of complexes, in which METTL3 and METTL 14 act as the active center of methyltransferases and WTAP, RBM15/15B, VIRMA and ZC3H13 play the part of adaptor proteins. In addition, METL16, HAKAI and ZCCHC4 were evidenced to be independently catalyzed the formation of m6A modification. FTO, ALKBH5 and ALKBH3 have been identified as m6A demethylases and they remove m6A marks on RNAs in an independent manner. Readers, involving biological functions by recognizing m6A modifications, include the YTH family, HNRNP family, IGFBPP1/2/3, EIF3 and ABCF1.
3. Mutual Regulatory Mechanisms between m6A Modification and miRNAs
Recently, a large number of studies have demonstrated that m6A modification on miRNAs play essential roles in various pathophysiological processes. Most interestingly, emerging evidence revealed that miRNAs also regulate m6A modification by altering expressions of m6A regulatory proteins [51,55][17][18]. So far, mutual regulatory mechanisms between m6A modification and miRNAs in multiple diseases have attracted a huge amount of attention.
As a series of noncoding and single-stranded small molecular RNA with a length of 18–24 nucleotides, miRNAs could target specific mRNA sites and promote degradation or inhibit translation of mRNA [56][19]. Although there are nearly 3000 miRNAs in mammals, the generation processes of different miRNAs are almost consistent. In detail, upon transcription from DNA, primary transcripts of miRNAs (pri-miRNAs) are spliced by RNase Ⅲ structure domain proteins and double-stranded RNA binding protein Drosha and Di George Syndrome critical region 8 (DGCR8) to form precursor of miRNAs (pre-miRNAs) in the nucleus. Then, pre-miRNAs are exported from nucleus into cytoplasm by forming a complex with a transporter protein exportin-5 and a GTP-binding nuclear protein Ran-GTP [53,54][20][21]. Once transported out of nucleus, pre-miRNAs are cleaved into mature miRNAs through another type III RNase Dicer. The mature miRNAs subsequently bind to mRNAs with the help of Ago proteins, thereby affecting the levels or translation processes of corresponding mRNAs [57][22]. Interestingly, advances in m6A modification in recent years have widely broadened mechanisms underlying miRNA processing and regulation. Specifically, emerging studies have shown that m6A modification and its regulatory proteins involve in the production of mature miRNAs, which in turn affect the level of m6A modification [58][23].
3.1. m
6
A Modifications Involves in miRNA Generation and Function
Current studies have indicated that m6A modification is involved in the generation process of miRNAs, thus affecting the level of mature miRNAs. Published results in the journal Nature in 2015 revealed that decreasing the level of m6A modification on pri-miRNAs by knocking down METTL3 expression could inhibit the binding of DGCR8 to pri-miRNAs, which led to about 70% miRNAs being downregulated by at least 30% [57][22]. Up to now, the mechanism according to which reduction of m6A modification on pri-miRNAs inhibits the maturation of miRNAs in a DGCR8-dependent manner has been found in different diseases. For example, catalyzed by over-expressed METTL3, high m6A modification can promote the maturity of miR-25 and miR-25-3p by strengthening the combination of DGCR8 and pri-miR-25 in pancreatic duct epithelial cells, and this may provoke malignant phenotype of pancreatic cancer cells [59][24]. The reduction level of m6A modification mediated by low expression of METTL3 and METTL14 makes the weaker recognition of pri-miR-126 by DGCR8, which hinders the maturation of miR-126, thereby activating the PI3K/AKT/mTOR pathway to promote the proliferation and activation of fibroblasts. Moreover, METTL3-dependent m6A was involved in the DGCR8-mediated maturation of pri-miR-126 in endometriosis development [60][25]. In addition, the interaction of METTL3 and DGCR8 positively modulates the biogenesis process of miR-873-5p, miR-365-3p and miR-221/222 in an m6A-dependent manner in different pathological processes, and as the simplest for specific miRNA, miR-873-5p participated in fighting colistin induced oxidative stress and apoptosis in kidney injury [61][26], miR-365-3p regulated chronic inflammatory pain induced by Complete Freund’s Adjuvant in the spinal cord [62][27], and miR-221/222 negatively mediate the PTEN expression, thus leading to the proliferation of bladder cancer cells [63][28]. In addition, cigarette smoke can stimulate the production of excess mature miRNA-93 in bronchial epithelial cells via enhanced m6A modification, which was mediated by overexpressed METTL3 [64][29]. METTL3 also plays a major catalytic role in m6A modification in unilateral ureteral obstruction mice and drove obstructive renal fibrosis development by promoting miR-21-5p maturation [65][30]. In addition, it has been indicated that silencing of METTL3 expression can elevate the levels of pri-miR-663 and m6A methylation-modified pri-miR-663, which resulted in suppressing of miR-663 maturation process in A549 and PC9LC cells [66][31]. In a manner similar to METTL3, the METTL14-mediated m6A marks also enhanced the recognition of pri-miR-126 by DGCR8, thus subsequently processing to mature miRNA-126, which is involved in hepatocellular carcinoma metastasis [60][25]. Different from the above mechanisms, METTL3 induced upregulation of miR-143-3p mostly depends on the shear effect of Dicer on pre-miR-143-3p in lung cancer cells [67][32]. Moreover, as in bone marrow-derived mesenchymal stem cells, METTL3 also methylate pre-miR-320, on which m6A modification is a key factor that is recognized and decayed by YTHDF2 [68][33]. METTL3 promoted the transition from pri-miR-1246 to mature miR-1246, of which upregulation can significantly enhance the metastasis ability of colorectal cancer cells [69][34]. METTL3-mediated m6A modification also promotes the expressions of 9 miRNAs, including miR-106b, miR-18a, miR-18b, miR-3607, miR-432, miR-30a, miR-320b, miR-320d and miR-320e, and bioinformatics analysis has shown that these miRNAs are involved in regulating signaling pathways closely related to malignant transformation induced by arsenite [70][35]. In addition, four miRNAs (miR-130a-3p, miR-130b-3p, miR-106b-5p and miR-301a-3p) are all related to short overall survival of kidney renal clear cell carcinoma patients and have significantly negative correlation with METTL14 mRNA [71][36]. Up to now, the importance of methyltransferases-catalyzed m6A modification on pri-miRNAs has been widely recognized, and a variety of methyltransferase components can affect the generation and function of miRNAs.
Besides methyltransferases, m6A demethylases and methyl-binding proteins are also involved in miRNA biogenesis. The earliest study showed that knocking down the FTO expression significantly increased levels of 42 miRNAs and decreased levels of 9 miRNAs [72][37]. A subsequent study reported that FTO regulates cell migration and invasion in breast cancer cells by inhibiting miR-181b-3p [73][38]. Moreover, FTO has been well evidenced to promoted bladder cancer cell proliferation via the FTO/miR-576/CDK6 pathways [74][39]. ALKBH5 inhibits tumor growth and metastasis by inhibiting miR-107/LATS2 mediated YAP activity in non-small cell lung cancer [75][40]. Peng et al. indicated that ALKBH5, the most potent member related to patient outcomes and to suppressing esophageal cancer malignancy in cell and animal models, demethylated pri-miR-194-2 and inhibited miR-194-2 biogenesis through an m6A/DGCR8-dependent manner [76][41]. Interestingly, in human non-small cell lung cancer cells, the depletion of ALKBH5 did not change the miR-21-5p level but altered the m6A abundance on miR-21-5p, thereby changing the miR-21-5p silencing potency towards its target mRNAs, which finally impaired the proliferation and motility of human non-small cell lung cancer cells [77][42]. In addition, ALKBH5 demethylated pri-miR-320a-3p, thus blocking DGCR8 from interacting with pri-miR-320a-3p and leading to mature process blockage of pri-miR-320a-3p in silica-inhaled mouse lung tissues [78][43].
In addition to being directly affected by m6A methyltransferases and demethylases, miRNA generation is also regulated by m6A methyl binding proteins. YTHDC1, a well-known m6A reader, facilitated the biogenesis of mature miR-30d via m6A-mediated regulation of mRNA stability. Furthermore, miR-30d represses pancreatic tumor genesis via suppressing aerobic glycolysis [79][44]. m6A reader protein HNRNPA2B1 also binds to a subset of m6A-modified pri-miRNA transcripts, thus interacting with DGCR8 and promoting primary miRNA processing, and depletion of HNRNPA2B1 caused a reduction in the levels of 61 miRNAs in HEK293 cells. Moreover, transiently overexpressed (5.4-fold) HNRNPA2B1 in MCF-7 cells led to significant alteration of more than 100 miRNAs, which regulate TGFβ and Notch signaling pathways according to MetaCore Enrichment analysis [80,81,82][45][46][47]. Yi et al. have reported that miR-185 transfer from vascular smooth muscle cells to endothelial cells is controlled by HNRNPA2B1 [83][48], but the role of m6A modification in this mediate process needs to be further explored. In addition, HNRNPA2B1 reads the m6A site on pri-miR-106b or pri-Let-7b to facilitate the maturing of miR-106b-5p or Let-7b in the lung cancer cells [84,85][49][50]. Another m6A binding protein, IGF2BP1, promotes serum response factor expression in an m6A-dependent manner by impairing the miRNA-directed downregulation of the serum response factor (SRF) mRNA in cancer cells [86][51]. In addition to regulating the generation process of miRNAs, m6A modification can directly modify mature miRNAs and affect their stability and degradation [72][37]. Of note, m6A modification on the E2F transcription factor 3 (E2F3) mRNA was required for the interaction between miR-660 and E2F3 mRNA in gastric cancer, indicating that m6A also affects the function of miRNA apart from participating in its production process [87][52].
In a word, emerging studies have identified the roles of m6A modification during the processing and maturation of miRNAs, which will surely provide good candidate targets for miRNA intervention. Although mechanisms underlying m6A modification affecting miRNA generation and function are diverse and complex, the general mechanisms are similar. The Weresearchers drew a schematic diagram, which take miR-30d [79][44], miR-21-5p [65][30] and pre-miR-25 [59][24] as examples, to show the specific mechanisms of m6A regulatory proteins regulating miRNAs (Figure 2).
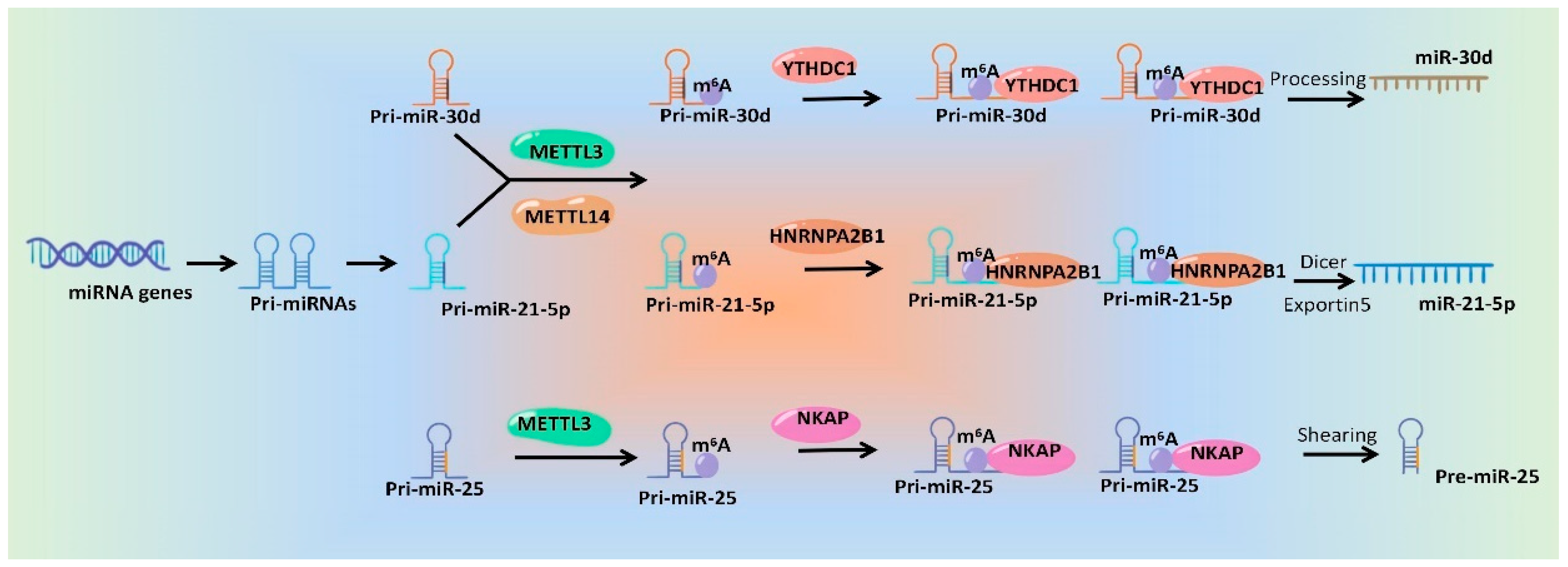
Figure 2.
Schematic diagram of the mechanisms of m
6
A modification and its regulatory proteins regulating miRNA production.
3.2. miRNAs Regulate the m
6
A Modification
Since miRNAs affect the protein level through interacting with mRNAs and m6A modification is a dynamic reversible methylation [88][53], it is rationality that miRNAs are involved in the regulation of m6A modification by affecting the regulatory proteins. At present, several studies have shown that miRNAs regulate m6A modification via sequence pairing of mRNAs of methyltransferases, demethylases, and methyl-binding proteins in various tissues [89][54]. In detail, METTL3 was identified as the direct target of miR-1269b [90][55] and miR-338-5p [91][56], thus inhibiting gastric cancer development. miR-33a is capable of reducing the METTL3 expression at both mRNA and protein levels, thus affecting proliferation, survival and invasion of non-small cell lung cancer [92][57]. Moreover, miR-600 can attenuate METTL3 expression and restrain the migration and proliferation of lung cancer cells [93][58]. Similarly, the down regulation of miR-524-5p also up-regulates the expression of METTL3 in non-small cell lung cancer cells [94][59]. miR-4429 targeted and repressed METTL3 to inhibit m6A-mediated stabilization of SEC62, a component belonging to tetrameric Sec62/Sec63-subcomplex of Sec-complex, thus hindering proliferation and encouraging apoptosis in gastric cancer cells [95][60]. Moreover, Cai et al. concluded that mammalian hepatitis B X-interacting protein (HBXIP) suppresses miRNA let-7g, thus up-regulating METTL3, which in turn promotes the expression of HBXIP through m6A modification, leading to stimulation or proliferation of breast cancer cells [96][61]. As an independent prognostic factor in hepatoblastoma patients, METTL3 was identified as a direct target of miR-186, of which low level led to high expression of METTL3, thus significantly inhibiting the proliferation, migration and invasion of hepatoblastoma cells [97][62]. Moreover, miR-320d has been evidenced to target METTL3, thus affecting KIF3C expression through changing m6A modification on KIF3C mRNA in prostate cancer cells [98][63]. Under the treatment of Mono-(2-ethylhexyl)phthalate (MEHP), miRNAs such as miR-16-1-3p, miR-101a-3p, miR-362-5p, miR-501-5p, miR-532-3p and miR-542-3p are dramatically activated in murine macrophage Raw 264.7 cells, and these miRNAs are all predicted to regulate METTL14, thus promoting m6A modification in Scavenger Receptor B type 1 (SR-B1) mRNA [99][64]. Cui et al. reported that miR-193a-3p directly targets ALKBH5 to inhibit the growth and promote the apoptosis of glioma cells by suppressing the AKT2 pathway both in vitro and in vivo [100][65]. Interestingly, circGPR137B acted as a sponge for miR-4739 to up-regulate its target FTO, which mediated m6A demethylation of circGPR137B and promoted its expression, thus finally forming a feedback loop comprising circGPR137B/miR-4739/FTO axis and affecting the hepatocellular carcinoma cells [101][66]. Results from Yang et al. indicated that imiR-155 directly targets FTO to negatively regulate its expression and increase m6A level in renal clear cell carcinoma cells. Regarding specific mechanisms, miR-155 is directly bound to the 3′-UTR of FTO mRNA and reduced FTO protein levels [102][67].
The methyl binding proteins of m6A modification are also directly targeted by miRNAs. In detail, Zheng et al. reported that miRNA-421-3p targets YTHDF1 to inhibit p65 mRNA translation, thus preventing inflammatory response in cerebral ischemia/reperfusion injury [103][68]. miR-376c also has been indicated to negatively modulate YTHDF1 expression in non-small cell lung cancer cells [104][69]. Negative correlations between the miR-145 level and YTHDF2 mRNA expression were observed in hepatocellular carcinoma [105][70] and epithelial ovarian cancer cells [106][71], and further detecting results showed that miR-145 decreased the luciferase activities of 3′-UTR of YTHDF2 mRNA, implicating that YTHDF2 is the direct target gene of miR-145 [105,106][70][71]. In addition, YTHDF2 mRNA is also regulated by miRNA-495 in prostate cancer cells [107][72] and miR-6125 in colorectal cancer cells [108][73]. Bioinformatics analysis from Hao et al. literature revealed IGF2BP1 as the putative target of miR-670, of which mimics and inhibitors were microinjected into parthenogenetic activation embryos, thus confirming these findings [109][74]. IGF2BP2, another m6A methyl binding protein, is highly expressed in thyroid cancer cells and identified as a target of miR-204 [110][75]. In addition, inhibition of miR-133b also resulted in the up regulation of IGF2BP2 in colorectal cancer cells [111][76]. Different from the above-mentioned mechanisms where miRNAs regulated m6A modification, results from Chen et al. indicated that overexpressing dicer increased the m6A modification level, and this was not achieved by alternating the quantity of m6A methyltransferases or demethylases in mouse embryonic fibroblasts. Further experiments showed that miRNAs regulate activity and location of METTL3, which subsequently modulate m6A modification and impede the reprogramming of mouse embryonic fibroblasts to pluripotent stem cells [112][77].
In a word, miRNAs can influence m6A modification by regulating the regulatory proteins of m6A and ultimately participate in a variety of pathological and physiological processes. WThe researchers drew a schematic diagram, in which wethe researchers take METTL3 [93][58], FTO [113][78] and YTHDF2 [108][73] as examples, to show the miRNAs involving in regulation of m6A modification and its biological effect (Figure 3).
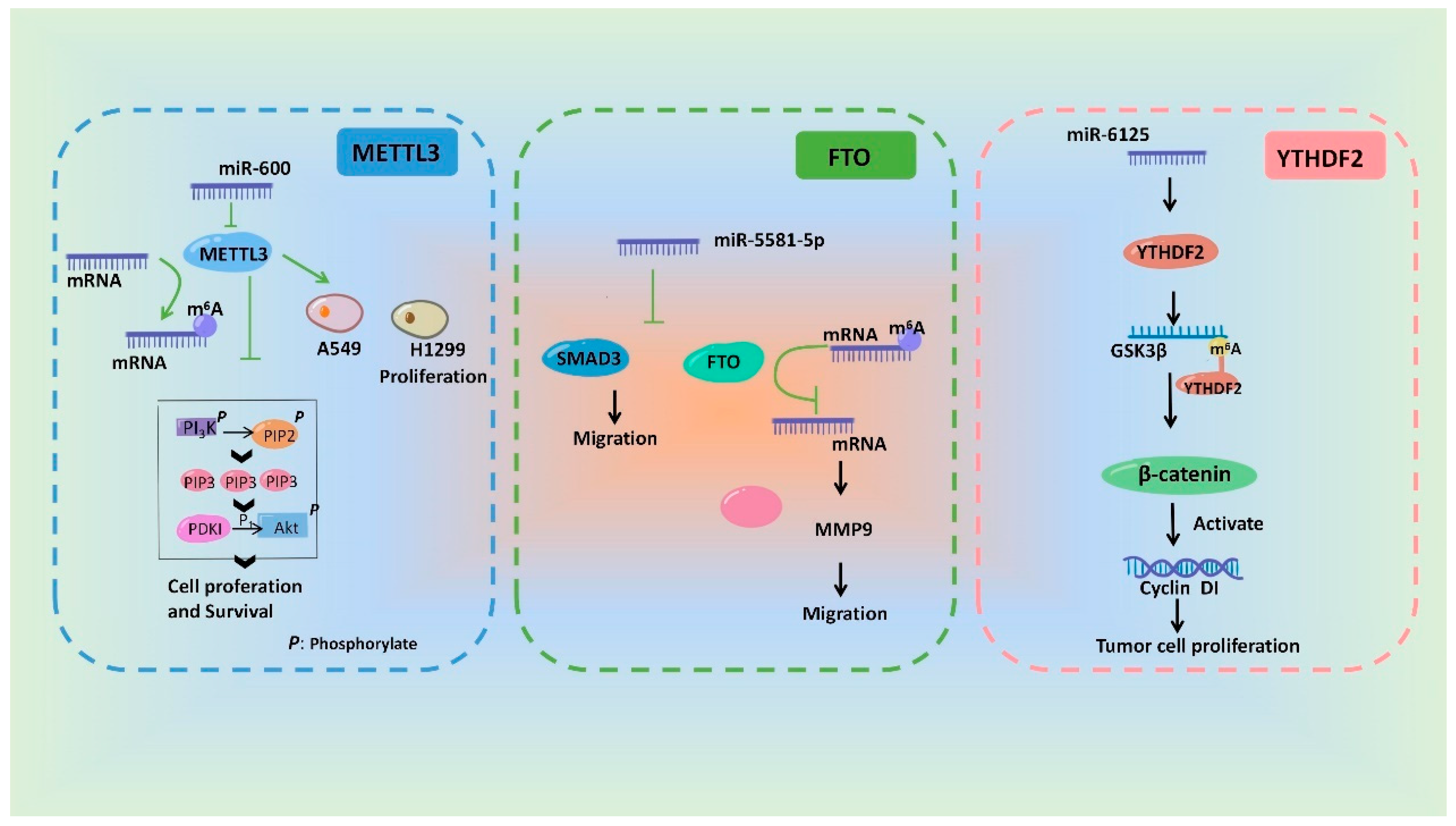
Figure 3.
Schematic diagram of the mechanisms showing that m
6
A modification and its biological effects are regulated by miRNAs.
As mentioned above, m6A modification is regulated by different m6A regulatory proteins in a variety of diseases by promoting biosynthesis of miRNAs, and miRNA regulates the biological functions of m6A regulatory proteins. Based on this interplay, wthe researchers summarized the change trends and regulation relationships of m6A regulatory proteins and miRNAs in tissues or cells during the occurrence and development of different diseases, as shown in Figure 4.
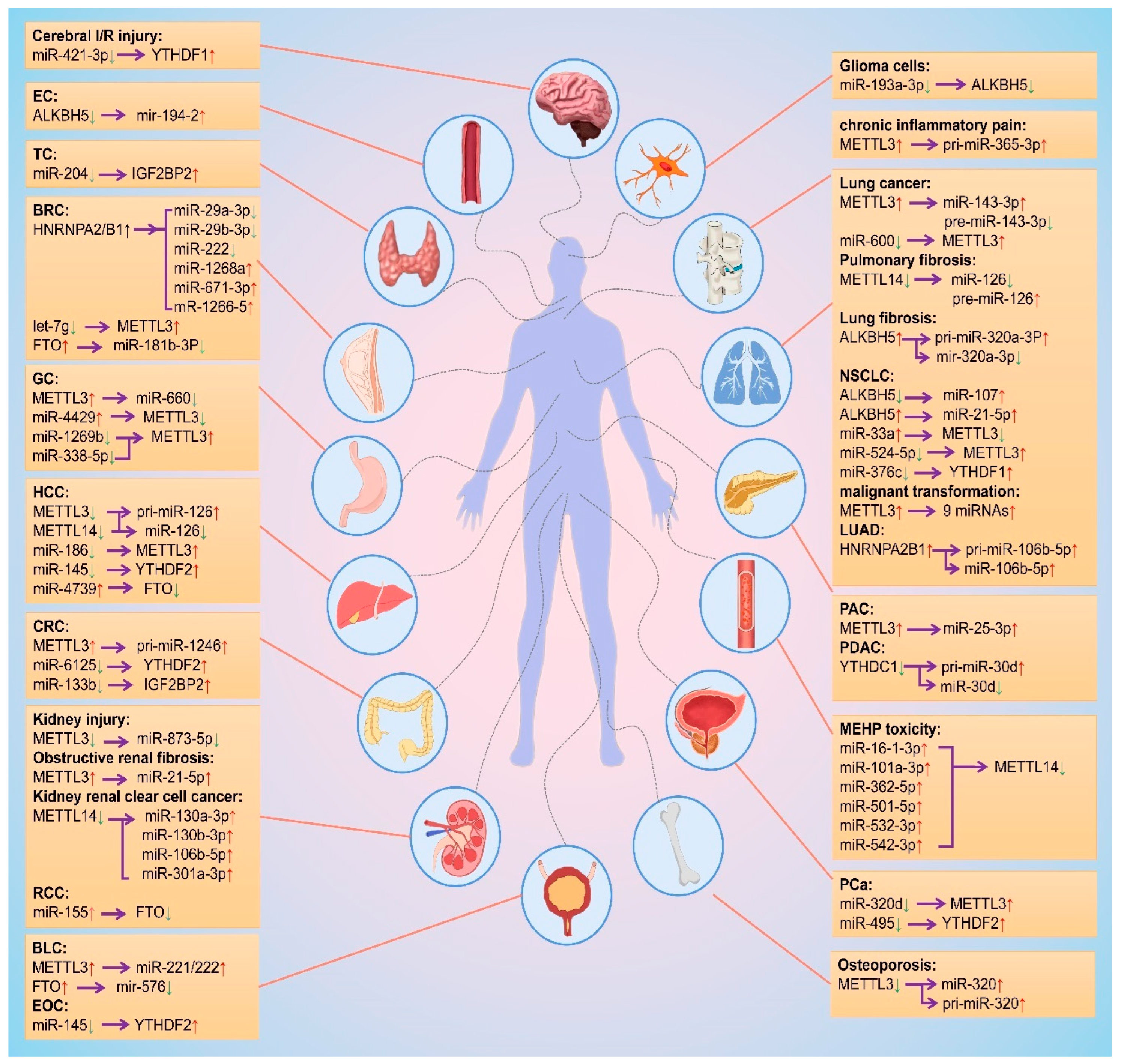
Figure 4. Mutual regulation between m6A modifications and miRNAs. The red and green arrows represent the level rise and fall, respectively. The purple arrows point to the regulated object. EC: esophageal cancer; TC: thyroid cancer; BRC: breast cancer; GC: gastric cancer; HCC: hepatocellular carcinoma; CRC: colorectal cancer; RCC: Renal cell carcinoma; BLC: bladder cancer; EOC: epithelial ovarian cancer; NSCLC: non-small cell lung cancer; LUAD: lung adenocarcinoma; PAC: pancreatic cancer; PDAC: Pancreatic ductal adenocarcinoma; PCa: prostate cancer.
References
- Xu, J.; Wan, Z.; Tang, M.; Lin, Z.; Jiang, S.; Ji, L.; Gorshkov, K.; Mao, Q.; Xia, S.; Cen, D.; et al. N6-methyladenosine-modified CircRNA-SORE sustains sorafenib resistance in hepatocellular carcinoma by regulating β-catenin signaling. Mol. Cancer 2020, 19, 163.
- Desrosiers, R.; Friderici, K.; Rottman, F. Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. Proc. Natl. Acad. Sci. USA 1974, 71, 3971–3975.
- Wang, S.; Lv, W.; Li, T.; Zhang, S.; Wang, H.; Li, X.; Wang, L.; Ma, D.; Zang, Y.; Shen, J.; et al. Dynamic regulation and functions of mRNA m6A modification. Cancer Cell Int. 2022, 22, 48.
- Yang, Y.; Hsu, P.J.; Chen, Y.S.; Yang, Y.G. Dynamic transcriptomic m6A decoration: Writers, erasers, readers and functions in RNA metabolism. Cell Res. 2018, 28, 616–624.
- Huang, H.; Weng, H.; Chen, J. The Biogenesis and Precise Control of RNA m6A Methylation. Trends Genet. 2020, 36, 44–52.
- Sun, T.; Wu, R.; Ming, L. The role of m6A RNA methylation in cancer. Biomed. Pharmacother. 2019, 112, 108613.
- Wei, W.; Ji, X.; Guo, X.; Ji, S. Regulatory Role of N6-methyladenosine (m6A) Methylation in RNA Processing and Human Diseases. J. Cell. Biochem. 2017, 118, 2534–2543.
- Zepecki, J.P.; Karambizi, D.; Fajardo, J.E.; Snyder, K.M.; Guetta-Terrier, C.; Tang, O.Y.; Chen, J.S.; Sarkar, A.; Fiser, A.; Toms, S.A.; et al. miRNA-mediated loss of m6A increases nascent translation in glioblastoma. PLoS Genet. 2021, 17, e1009086.
- Wiener, D.; Schwartz, S. The epitranscriptome beyond m6A. Nat. Rev. Genet. 2021, 22, 119–131.
- Lin, C.; Ma, M.; Zhang, Y.; Li, L.; Long, F.; Xie, C.; Xiao, H.; Liu, T.; Tian, B.; Yang, K.; et al. Correction to: The N6-methyladenosine modification of circALG1 promotes the metastasis of colorectal cancer mediated by the miR-342-5p/PGF signalling pathway. Mol. Cancer 2022, 21, 80.
- Khan, A.; Rehman, H.U.; Habib, U.; Ijaz, U. m6A-Finder: Detecting m6A methylation sites from RNA transcriptomes using physical and statistical properties based features. Comput. Biol. Chem. 2022, 97, 107640.
- Meyer, K.D.; Saletore, Y.; Zumbo, P.; Elemento, O.; Mason, C.E.; Jaffrey, S.R. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell 2012, 149, 1635–1646.
- Zhang, B.; Chen, Z.; Tao, B.; Yi, C.; Lin, Z.; Li, Y.; Shao, W.; Lin, J.; Chen, J. m6A target microRNAs in serum for cancer detection. Mol. Cancer 2021, 20, 170.
- Yi, Y.; Chen, X.; Zhang, J.; Zhu, J. Novel insights into the interplay between m6A modification and noncoding RNAs in cancer. Mol. Cancer 2020, 19, 121.
- Tang, F.; Chen, L.; Gao, H.; Xiao, D.; Li, X. m6A: An Emerging Role in Programmed Cell Death. Front. Cell Dev. Biol. 2022, 10, 817112.
- Sun, Z.; Wang, H.; Wang, Y.; Yuan, G.; Yu, X.; Jiang, H.; Wu, Q.; Yang, B.; Hu, Z.; Shi, F.; et al. MiR-103-3p targets the m6A methyltransferase METTL14 to inhibit osteoblastic bone formation. Aging Cell 2021, 20, e13298.
- Zhou, K.I.; Parisien, M.; Dai, Q.; Liu, N.; Diatchenko, L.; Sachleben, J.R.; Pan, T. N6-Methyladenosine Modification in a Long Noncoding RNA Hairpin Predisposes Its Conformation to Protein Binding. J. Mol. Biol. 2016, 428, 822–833.
- Dai, D.; Wang, H.; Zhu, L.; Jin, H.; Wang, X. N6-methyladenosine links RNA metabolism to cancer progression. Cell Death Dis. 2018, 9, 113–124.
- Afonso-Grunz, F.; Müller, S. Principles of miRNA–mRNA interactions: Beyond sequence complementarity. Cell. Mol. Life Sci. 2015, 72, 3127–3141.
- Liu, N.; Zhou, K.I.; Parisien, M.; Dai, Q.; Diatchenko, L.; Pan, T. N 6-methyladenosine alters RNA structure to regulate binding of a low-complexity protein. Nucleic Acids Res. 2017, 45, 6051–6063.
- Edupuganti, R.R.; Geiger, S.; Lindeboom, R.G.H.; Shi, H.; Hsu, P.J.; Lu, Z.; Wang, S.; Baltissen, M.P.A.; Jansen, P.W.T.C.; Rossa, M.; et al. N6-methyladenosine (m6A) recruits and repels proteins to regulate mRNA homeostasis. Nat. Struct. Mol. Biol. 2017, 24, 870–878.
- Alarcón, C.R.; Lee, H.; Goodarzi, H.; Halberg, N.; Tavazoie, S.F. N6-methyladenosine marks primary microRNAs for processing. Nature 2015, 519, 482–485.
- Saliminejad, K.; Khorram Khorshid, H.R.; Soleymani Fard, S.; Ghaffari, S.H. An overview of microRNAs: Biology, functions, therapeutics, and analysis methods. J. Cell. Physiol. 2019, 234, 5451–5465.
- Zhang, J.; Bai, R.; Li, M.; Ye, H.; Wu, C.; Wang, C.; Li, S.; Tan, L.; Mai, D.; Li, G.; et al. Excessive miR-25-3p maturation via N6-methyladenosine stimulated by cigarette smoke promotes pancreatic cancer progression. Nat. Commun. 2019, 10, 1858.
- Zailaie, S.A.; Sergi, C.M. MiR-126 in Hepatocellular Carcinoma and Cholangiocellular Carcinoma: A Reappraisal with an in situ Detection of miR-126. Ann. Clin. Lab. Sci. 2022, 52, 73–85.
- Wang, J.; Ishfaq, M.; Xu, L.; Xia, C.; Chen, C.; Li, J. METTL3/m6A/miRNA-873-5p Attenuated Oxidative Stress and Apoptosis in Colistin-Induced Kidney Injury by Modulating Keap1/Nrf2 Pathway. Front. Pharmacol. 2019, 10, 517.
- Zhang, C.; Wang, Y.; Peng, Y.; Xu, H.; Zhou, X. METTL3 regulates inflammatory pain by modulating m6A-dependent pri-miR-365-3p processing. FASEB J. 2020, 34, 122–132.
- Han, J.; Wang, J.; Yang, X.; Yu, H.; Zhou, R.; Lu, H.; Yuan, W.; Lu, J.; Zhou, Z.; Lu, Q.; et al. METTL3 promote tumor proliferation of bladder cancer by accelerating pri-miR221/222 maturation in m6A-dependent manner. Mol. Cancer 2019, 18, 110.
- Wang, Z.; Sun, W.; Li, R.; Liu, Y. miRNA-93-5p in exosomes derived from M2 macrophages improves lipopolysaccharide-induced podocyte apoptosis by targeting Toll-like receptor 4. Bioengineered 2022, 13, 7683–7696.
- Liu, E.; Lv, L.; Zhan, Y.; Ma, Y.; Feng, J.; He, Y.; Wen, Y.; Zhang, Y.; Pu, Q.; Ji, F.; et al. METTL3/N6-methyladenosine/ miR-21-5p promotes obstructive renal fibrosis by regulating inflammation through SPRY1/ERK/NF-κB pathway activation. J. Cell. Mol. Med. 2021, 25, 7660–7674.
- Li, S.; Lu, X.; Zheng, D.; Chen, W.; Li, Y.; Li, F. Methyltransferase-like 3 facilitates lung cancer progression by accelerating m6A methylation-mediated primary miR-663 processing and impeding SOCS6 expression. J. Cancer Res. Clin. Oncol. 2022, 148, 3485–3499.
- Wang, H.; Deng, Q.; Lv, Z.; Ling, Y.; Hou, X.; Chen, Z.; Dinglin, X.; Ma, S.; Li, D.; Wu, Y.; et al. N6-methyladenosine induced miR-143-3p promotes the brain metastasis of lung cancer via regulation of VASH1. Mol. Cancer 2019, 18, 181.
- Yan, G.; Yuan, Y.; He, M.; Gong, R.; Lei, H.; Zhou, H.; Wang, W.; Du, W.; Ma, T.; Liu, S.; et al. m6A Methylation of Precursor-miR-320/RUNX2 Controls Osteogenic Potential of Bone Marrow-Derived Mesenchymal Stem Cells. Mol. Ther. Nucleic Acids 2020, 19, 421–436.
- Peng, W.; Li, J.; Chen, R.; Gu, Q.; Yang, P.; Qian, W.; Ji, D.; Wang, Q.; Zhang, Z.; Tang, J.; et al. Upregulated METTL3 promotes metastasis of colorectal Cancer via miR-1246/SPRED2/MAPK signaling pathway. J. Exp. Clin. Cancer Res. 2019, 38, 393.
- Gu, S.; Sun, D.; Dai, H.; Zhang, Z. N6-methyladenosine mediates the cellular proliferation and apoptosis via microRNAs in arsenite-transformed cells. Toxicol. Lett. 2018, 292, 1–11.
- Wang, Q.; Zhang, H.; Chen, Q.; Wan, Z.; Gao, X.; Qian, W. Identification of METTL14 in Kidney Renal Clear Cell Carcinoma Using Bioinformatics Analysis. Dis. Markers 2019, 2019, 5648783.
- Berulava, T.; Rahmann, S.; Rademacher, K.; Klein-Hitpass, L.; Horsthemke, B. N6-Adenosine Methylation in MiRNAs. PLoS ONE 2015, 10, e118438.
- Xu, Y.; Ye, S.; Zhang, N.; Zheng, S.; Liu, H.; Zhou, K.; Wang, L.; Cao, Y.; Sun, P.; Wang, T. The FTO/miR-181b-3p/ARL5B signaling pathway regulates cell migration and invasion in breast cancer. Cancer Commun. 2020, 40, 484–500.
- Zhou, G.; Yan, K.; Liu, J.; Gao, L.; Jiang, X.; Fan, Y. FTO promotes tumour proliferation in bladder cancer via the FTO/miR-576/CDK6 axis in an m6A-dependent manner. Cell Death Discov. 2021, 7, 329.
- Jin, D.; Guo, J.; Wu, Y.; Yang, L.; Wang, X.; Du, J.; Dai, J.; Chen, W.; Gong, K.; Miao, S.; et al. m6A demethylase ALKBH5 inhibits tumor growth and metastasis by reducing YTHDFs-mediated YAP expression and inhibiting miR-107/LATS2–mediated YAP activity in NSCLC. Mol. Cancer 2020, 19, 40.
- Chen, P.; Li, S.; Zhang, K.; Zhao, R.; Cui, J.; Zhou, W.; Liu, Y.; Zhang, L.; Cheng, Y. N6-methyladenosine demethylase ALKBH5 suppresses malignancy of esophageal cancer by regulating microRNA biogenesis and RAI1 expression. Oncogene 2021, 40, 5600–5612.
- Wang, H.; Song, X.; Song, C.; Wang, X.; Cao, H. m6A-seq analysis of microRNAs reveals that the N6-methyladenosine modification of miR-21–5p affects its target expression. Arch. Biochem. Biophys. 2021, 711, 109023.
- Sun, W.; Li, Y.; Ma, D.; Liu, Y.; Xu, Q.; Cheng, D.; Li, G.; Ni, C. ALKBH5 promotes lung fibroblast activation and silica-induced pulmonary fibrosis through miR-320a-3p and FOXM1. Cell Mol. Biol. Lett. 2022, 27, 26.
- Hou, Y.; Zhang, Q.; Pang, W.; Hou, L.; Liang, Y.; Han, X.; Luo, X.; Wang, P.; Zhang, X.; Li, L.; et al. YTHDC1-mediated augmentation of miR-30d in repressing pancreatic tumorigenesis via attenuation of RUNX1-induced transcriptional activation of Warburg effect. Cell Death Differ. 2021, 28, 3105–3124.
- Jiang, L.; Lin, W.; Zhang, C.; Ash, P.E.A.; Verma, M.; Kwan, J.; van Vliet, E.; Yang, Z.; Cruz, A.L.; Boudeau, S.; et al. Interaction of tau with HNRNPA2B1 and N6-methyladenosine RNA mediates the progression of tauopathy. Mol. Cell 2021, 81, 4209–4227.
- Klinge, C.M.; Piell, K.M.; Tooley, C.S.; Rouchka, E.C. HNRNPA2/B1 is upregulated in endocrine-resistant LCC9 breast cancer cells and alters the miRNA transcriptome when overexpressed in MCF-7 cells. Sci. Rep. 2019, 9, 9430.
- Luo, K. Signaling Cross Talk between TGF-beta/Smad and Other Signaling Pathways. Cold Spring Harb. Perspect. Biol. 2017, 9, a022137.
- Si, Y.; Liu, F.; Wang, D.; Fang, C.; Tang, X.; Guo, B.; Shi, Z.; Dong, Z.; Guo, D.; Yue, J.; et al. Exosomal Transfer of miR-185 Is Controlled by hnRNPA2B1 and Impairs Re-endothelialization After Vascular Injury. Front. Cell Dev. Biol. 2021, 9, 619444.
- Rong, L.; Xu, Y.; Zhang, K.; Jin, L.; Liu, X. HNRNPA2B1 inhibited SFRP2 and activated Wnt-beta/catenin via m6A-mediated miR-106b-5p processing to aggravate stemness in lung adenocarcinoma. Pathol. Res. Pract. 2022, 233, 153794.
- Li, K.; Gao, S.; Ma, L.; Sun, Y.; Peng, Z.; Wu, J.; Du, N.; Ren, H.; Tang, S.; Sun, X. Stimulation of Let-7 Maturation by Metformin Improved the Response to Tyrosine Kinase Inhibitor Therapy in an m6A Dependent Manner. Front. Oncol. 2022, 11, 731561.
- Müller, S.; Glaß, M.; Singh, A.K.; Haase, J.; Bley, N.; Fuchs, T.; Lederer, M.; Dahl, A.; Huang, H.; Chen, J.; et al. IGF2BP1 promotes SRF-dependent transcription in cancer in a m6A- and miRNA-dependent manner. Nucleic Acids Res. 2019, 47, 375–390.
- He, X.; Shu, Y. RNA N6-methyladenosine modification participates in miR-660/E2F3 axis-mediated inhibition of cell proliferation in gastric cancer. Pathol.-Res. Pract. 2019, 215, 152393.
- Chen, Y.; Lin, Y.; Shu, Y.; He, J.; Gao, W. Interaction between N6-methyladenosine (m6A) modification and noncoding RNAs in cancer. Mol. Cancer 2020, 19, 84.
- Ma, J.Z.; Yang, F.; Zhou, C.C.; Liu, F.; Yuan, J.H.; Wang, F.; Wang, T.T.; Xu, Q.G.; Zhou, W.P.; Sun, S.H. METTL14 suppresses the metastatic potential of hepatocellular carcinoma by modulating N6 -methyladenosine-dependent primary MicroRNA processing. Hepatology 2017, 65, 529–543.
- Kang, J.; Huang, X.; Dong, W.; Zhu, X.; Li, M.; Cui, N. MicroRNA-1269b inhibits gastric cancer development through regulating methyltransferase-like 3 (METTL3). Bioengineered 2021, 12, 1150–1160.
- Wang, G.; Zhang, Z.; Xia, C. Long non-coding RNA LINC00240 promotes gastric cancer progression via modulating miR-338-5p/METTL3 axis. Bioengineered 2021, 12, 9678–9691.
- Du, M.; Zhang, Y.; Mao, Y.; Mou, J.; Zhao, J.; Xue, Q.; Wang, D.; Huang, J.; Gao, S.; Gao, Y. MiR-33a suppresses proliferation of NSCLC cells via targeting METTL3 mRNA. Biochem. Biophys. Res. Commun. 2017, 482, 582–589.
- Wei, W.; Huo, B.; Shi, X. miR-600 inhibits lung cancer via downregulating the expression of METTL3. Cancer Manag. Res. 2019, 11, 1177–1187.
- Xie, H.; Yao, J.; Wang, Y.; Ni, B. Exosome-transmitted circVMP1 facilitates the progression and cisplatin resistance of non-small cell lung cancer by targeting miR-524-5p-METTL3/SOX2 axis. Drug Deliv. 2022, 29, 1257–1271.
- He, H.; Wu, W.; Sun, Z.; Chai, L. MiR-4429 prevented gastric cancer progression through targeting METTL3 to inhibit m6A-caused stabilization of SEC62. Biochem. Biophys. Res. Commun. 2019, 517, 581–587.
- Cai, X.; Wang, X.; Cao, C.; Gao, Y.; Zhang, S.; Yang, Z.; Liu, Y.; Zhang, X.; Zhang, W.; Ye, L. HBXIP-elevated methyltransferase METTL3 promotes the progression of breast cancer via inhibiting tumor suppressor let-7g. Cancer Lett. 2018, 415, 11–19.
- Cui, X.; Wang, Z.; Li, J.; Zhu, J.; Ren, Z.; Zhang, D.; Zhao, W.; Fan, Y.; Zhang, D.; Sun, R. Cross talk between RNA N6-methyladenosine methyltransferase-like 3 and miR-186 regulates hepatoblastoma progression through Wnt/β-catenin signalling pathway. Cell Prolif. 2020, 53, e12768.
- Ma, H.; Zhang, F.; Zhong, Q.; Hou, J. METTL3-mediated m6A modification of KIF3C-mRNA promotes prostate cancer progression and is negatively regulated by miR-320d. Aging 2021, 13, 22332–22344.
- Park, M.H.; Jeong, E.; Choudhury, M. Mono-(2-Ethylhexyl)phthalate Regulates Cholesterol Efflux via MicroRNAs Regulated m6A RNA Methylation. Chem. Res. Toxicol. 2020, 33, 461–469.
- Cui, Y.; Wang, Q.; Lin, J.; Zhang, L.; Zhang, C.; Chen, H.; Qian, J.; Luo, C. miRNA-193a-3p Regulates the AKT2 Pathway to Inhibit the Growth and Promote the Apoptosis of Glioma Cells by Targeting ALKBH5. Front. Oncol. 2021, 11, 600451.
- Liu, L.; Gu, M.; Ma, J.; Wang, Y.; Li, M.; Wang, H.; Yin, X.; Li, X. CircGPR137B/miR-4739/FTO feedback loop suppresses tumorigenesis and metastasis of hepatocellular carcinoma. Mol. Cancer 2022, 21, 149.
- Yang, W.; Xie, L.; Wang, P.; Zhuang, C. MiR-155 regulates m6A level and cell progression by targeting FTO in clear cell renal cell carcinoma. Cell. Signal. 2022, 91, 110217.
- Zheng, L.; Tang, X.; Lu, M.; Sun, S.; Xie, S.; Cai, J.; Zan, J. microRNA-421-3p prevents inflammatory response in cerebral ischemia/reperfusion injury through targeting m6A Reader YTHDF1 to inhibit p65 mRNA translation. Int. Immunopharmacol. 2020, 88, 106937.
- Zhou, J.; Xiao, D.; Qiu, T.; Li, J.; Liu, Z. Loading MicroRNA-376c in Extracellular Vesicles Inhibits Properties of Non-Small Cell Lung Cancer Cells by Targeting YTHDF1. Technol. Cancer Res. Treat. 2020, 19, 1180565672.
- Yang, Z.; Li, J.; Feng, G.; Gao, S.; Wang, Y.; Zhang, S.; Liu, Y.; Ye, L.; Li, Y.; Zhang, X. MicroRNA-145 Modulates N6-Methyladenosine Levels by Targeting the 3′-Untranslated mRNA Region of the N6-Methyladenosine Binding YTH Domain Family 2 Protein. J. Biol. Chem. 2017, 292, 3614–3623.
- Li, J.; Wu, L.; Pei, M.; Zhang, Y. YTHDF2, a protein repressed by miR-145, regulates proliferation, apoptosis, and migration in ovarian cancer cells. J. Ovarian Res. 2020, 13, 111.
- Du, C.; Lv, C.; Feng, Y.; Yu, S. Activation of the KDM5A/miRNA-495/YTHDF2/m6A-MOB3B axis facilitates prostate cancer progression. J. Exp. Clin. Cancer Res. 2020, 39, 223.
- Li, H.; Zhang, N.; Jiao, X.; Wang, C.; Sun, W.; He, Y.; Ren, G.; Huang, S.; Li, M.; Chang, Y.; et al. Downregulation of microRNA-6125 promotes colorectal cancer growth through YTHDF2-dependent recognition of N6-methyladenosine-modified GSK3β. Clin. Transl. Med. 2021, 11, e602.
- Hao, J.; Hu, H.; Jiang, Z.; Yu, X.; Li, C.; Chen, L.; Xia, Y.; Liu, D.; Wang, D. microRNA-670 modulates Igf2bp1 expression to regulate RNA methylation in parthenogenetic mouse embryonic development. Sci. Rep. 2020, 10, 4782.
- Ye, M.; Dong, S.; Hou, H.; Zhang, T.; Shen, M. Oncogenic Role of Long Noncoding RNAMALAT1 in Thyroid Cancer Progression through Regulation of the miR-204/IGF2BP2/m6A-MYC Signaling. Mol. Ther.-Nucleic Acids 2021, 23, 1–12.
- Yao, B.; Zhang, Q.; Yang, Z.; An, F.; Nie, H.; Wang, H.; Yang, C.; Sun, J.; Chen, K.; Zhou, J.; et al. CircEZH2/miR-133b/IGF2BP2 aggravates colorectal cancer progression via enhancing the stability of m6A-modified CREB1 mRNA. Mol. Cancer 2022, 21, 140.
- Chen, T.; Hao, Y.; Zhang, Y.; Li, M.; Wang, M.; Han, W.; Wu, Y.; Lv, Y.; Hao, J.; Wang, L.; et al. m6A RNA Methylation Is Regulated by MicroRNAs and Promotes Reprogramming to Pluripotency. Cell Stem Cell 2015, 16, 289–301.
- Sun, J.; Ma, X.; Ying, Y.; Wang, W.; Shen, H.; Wang, S.; Xie, H.; Yi, J.; Zhan, W.; Li, J.; et al. SMAD3 and FTO are involved in miR-5581-3p-mediated inhibition of cell migration and proliferation in bladder cancer. Cell Death Discov. 2022, 8, 199.
More