Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Conner Chen and Version 1 by pan xinhui.
Isoxazoles and isoxazolines are five-membered heterocyclic molecules containing nitrogen and oxygen. Isoxazole and isoxazoline are the most popular heterocyclic compounds for developing novel drug candidates. As biosynthetic technology has advanced, A growing number of natural products are being developed for cancer therapy as clinical candidates. As a result, scientists have created several isoxazole and isoxazoline derivatives with anti-cancer properties based on natural products.
- isoxazole
- isoxazoline
- natural products
1. Introduction
Isoxazole is a heterocyclic compound with a five-membered ring that has oxygen and nitrogen atoms at the 1 and 2 positions, and their partially saturated analogs are known as isoxazoline. Many biologically active products contain derivatives of these heterocyclic compounds [1,2,3][1][2][3]. Derivatives containing isoxazole/isoxazoline fragments possess biological activities such as anticancer [4[4][5],5], anti-inflammatory [6[6][7],7], antibacterial [8[8][9],9], anti-Alzheimer’s disease [10[10][11],11], antioxidant [12[12][13],13], insecticidal [14], antifungal [15,16][15][16], and antidiabetic [17,18][17][18]. Isoxazolines and isoxazoles have unique electron-rich aromatic structures and have received much attention [19,20][19][20]. They make potential candidates for ring cleavage because of their weak nitrogen–oxygen bonds and aromatic character. Thus, isoxazoles and isoxazolines are particularly valuable intermediates in numerous synthetic methods of bioactive chemicals because this isoxazole ring system makes it easy to modify the substituents in their ring structures [5]. Their unusual architectures enable high-affinity binding to many targets or multiple distinct receptors, which aids in the development of innovative medications with original therapeutic applications. Therefore, chemists have been interested in developing and testing isoxazole- and isoxazoline-containing compounds with a variety of medicinal benefits [21].
In the drug discovery process, natural products play a significant role. Biologically active natural products can be obtained from plants, marine organisms, or microorganisms, and they are a vital source of drug discovery [22]. However, most directly extracted and isolated unmodified natural products cannot be directly used for clinical treatment of diseases due to the low pharmacological activity or excessive adverse effects of most natural products. Structural modifications can be used to improve the physicochemical properties and biological activity to reduce the adverse effects and improve the drug selectivity of natural products. They may also exhibit completely different biological activities from the parent [23]. Given the importance of the isoxazole/isoxazoline backbone in natural products and the remarkable bioactivity, an increasing number of studies have reported the modification of natural products by isoxazole/isoxazoline rings [24].
2. Antitumour Activity
Malignant tumors are extremely complex, and they can have a major impact on people’s life and health. The incidence of cancer has been on the rise, and despite the availability of many drugs and treatments for cancer, it remains one of the greatest threats to human health. Natural products or their derivatives make up over 65% of all anti-cancer medications. Natural products thus serve a crucial clinical role in the treatment of cancer. As biosynthetic technology has advanced, A growing number of natural products are being developed for cancer therapy as clinical candidates [25]. As a result, scientists have created several isoxazole and isoxazoline derivatives with anti-cancer properties based on natural products. Maslinic acid (MA)(Figure 1) and oleanolic acid (OA) (Figure 2) can be isolated from the natural Olea europaea L. MA as well as OA have been found to have anti-cancer and anti-inflammatory properties. A number of isoxazole-containing pentacyclic triterpene derivatives were created and examined by Chouaïb and coworkers. The majority of the isoxazoles, especially those generated from MA, showed remarkable anti-cancer activity in tests on the cancer cell lines EMT-6 and SW480. Isoxazole derivatives of MA 1a, 1b, 1c, and 1d (Figure 1) showed better anti-cancer properties against SW480 cell line compared to the starting substrate MA (viability (%/control) = 9, 9, 10, and 10%, respectively, 30 µM). However, only compounds 1d and 1c showed higher activity than MA against EMT6 (breast) (viability (%/control) = 5 and 64%, respectively, 10 µM). The scientists also performed an anti-proliferative assessment on the cancer cell lines EMT-6 and SW480 and documented the synthesis of OA isoxazole derivatives. However, according to in vitro cytotoxicity testing, OA contains a more potent anti-proliferative active compound than its isoxazole derivatives [26]. In another study, A number of OA nitrogen heterocyclic derivatives with nitrogen heterocycles at C-2 and C-3 were created by Mallavadhani et al. According to the study, pyrimidine derivatives had much greater activity than isoxazoles derivative 2 (Figure 2) against seven cancerous cell lines. The pyrimidine derivatives stopped the cell cycle and caused apoptosis in MCF cells during the S phase, according to the flow cytometric study [27].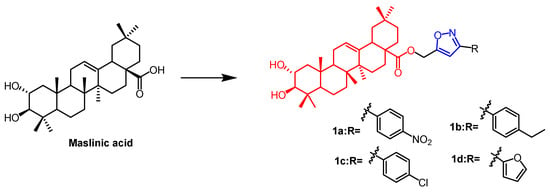
Figure 1. The chemical structure and derivative of maslinic. (The red marker in the figure indicates the parent structure of MA, and the blue marker indicates the structural modification of isoxazole.) [26].
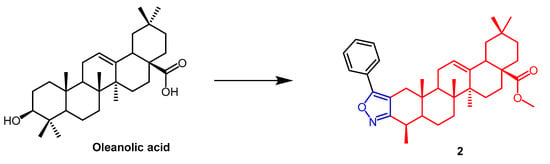
Figure 2. The chemical structure and derivatives of oleanolic. (The red marker in the figure indicates the parent structure of OA, and the blue marker indicates the structural modification of isoxazole.) The same explanation for the following figures [27].
References
- Kumar, G.; Shankar, R. 2-Isoxazolines: A Synthetic and Medicinal Overview. Chemmedchem 2021, 16, 430–447.
- Pandhurnekar, C.P.; Pandhurnekar, H.C.; Mungole, A.J.; Butoliya, S.S.; Yadao, B.G. A review of recent synthetic strategies and biological activities of isoxazole. J. Heterocycl. Chem. 2022, 60, 1–29.
- Tilvi, S.; Singh, K.S. Synthesis of Oxazole, Oxazoline and Isoxazoline Derived Marine Natural Products: A Review. Curr. Org. Chem. 2016, 20, 898–929.
- Arya, G.C.; Kaur, K.; Jaitak, V. Isoxazole derivatives as anticancer agent: A review on synthetic strategies, mechanism of action and SAR studies. Eur. J. Med. Chem. 2021, 221, 113511.
- Kaur, K.; Kumar, V.; Sharma, A.K.; Gupta, G.K. Isoxazoline containing natural products as anticancer agents: A review. Eur. J. Med. Chem. 2014, 77, 121–133.
- Abu-Hashem, A.A.; El-Shazly, M. Synthesis of New Isoxazole-, Pyridazine-, Pyrimidopyrazines and their Anti-Inflammatory and Analgesic Activity. Med. Chem. 2018, 14, 356–371.
- Mota, F.V.B.; Neta, M.S.D.; Franco, E.D.; Bastos, I.; da Araujo, L.C.C.; da Silva, S.C.; de Oliveira, T.B.; Souza, E.K.; de Almeida, V.M.; Ximenes, R.M.; et al. Evaluation of anti-inflammatory activity and molecular docking study of new aza-bicyclic isoxazoline acylhydrazone derivatives. Medchemcomm 2019, 10, 1916–1925.
- Aarjane, M.; Slassi, S.; Ghaleb, A.; Tazi, B.; Amine, A. Synthesis, biological evaluation, molecular docking and in silico ADMET screening studies of novel isoxazoline derivatives from acridone. Arab. J. Chem. 2021, 14, 103057.
- Shaik, A.; Bhandare, R.R.; Palleapati, K.; Nissankararao, S.; Kancharlapalli, V.; Shaik, S. Antimicrobial, Antioxidant, and Anticancer Activities of Some Novel Isoxazole Ring Containing Chalcone and Dihydropyrazole Derivatives. Molecules 2020, 25, 1047.
- Rastegari, A.; Safavi, M.; Vafadarnejad, F.; Najafi, Z.; Hariri, R.; Bukhari, S.N.A.; Iraji, A.; Edraki, N.; Firuzi, O.; Saeedi, M.; et al. Synthesis and evaluation of novel arylisoxazoles linked to tacrine moiety: In vitro in vivo biological activities against Alzheimer’s disease. Mol. Divers. 2022, 26, 409–428.
- Patil, P.; Thakur, A.; Sharma, A.; Flora, S.J.S. Natural products and their derivatives as multifunctional ligands against Alzheimer’s disease. Drug Dev. Res. 2020, 81, 165–183.
- Gul, M.; Eryilmaz, S. Synthesis, Antioxidant Activity and Theoretical Investigation of Isoxazolines Derivatives of Monoterpenoids. Lett. Org. Chem. 2019, 16, 501–510.
- Pothuri, V.V.; Machiraju, P.V.S.; Rao, V.S.S. Synthesis and Biological Activity of Some Novel Derivatives of 4-dioxin-7-yl)isoxazole-3-yl]benzoic Acid. Russ. J. Gen. Chem. 2020, 90, 889–894.
- Huang, S.S.; Zhu, B.B.; Wang, K.H.; Yu, M.; Wang, Z.W.; Li, Y.Q.; Liu, Y.X.; Zhang, P.L.; Li, S.J.; Li, Y.L.; et al. Design, synthesis, and insecticidal and fungicidal activities of quaternary ammonium salt derivatives of a triazolyphenyl isoxazoline insecticide. Pest Manag. Sci. 2022, 78, 2011–2021.
- Trefzger, O.S.; Barbosa, N.V.; Scapolatempo, R.L.; das Neves, A.R.; Ortale, M.; Carvalho, D.B.; Honorato, A.M.; Fragoso, M.R.; Shuiguemoto, C.Y.K.; Perdomo, R.T.; et al. Design, synthesis, antileishmanial, and antifungal biological evaluation of novel 3,5-disubstituted isoxazole compounds based on 5-nitrofuran scaffolds. Arch. Der Pharm. 2020, 353, 1900241.
- Zhang, T.; Dong, M.Y.; Zhao, J.J.; Zhang, X.F.; Mei, X.D. Synthesis and antifungal activity of novel pyrazolines and isoxazolines derived from cuminaldehyde. J. Pestic. Sci. 2019, 44, 181–185.
- Li, Z.; Liu, C.X.; Shi, W.; Cai, X.G.; Dai, Y.X.; Liao, C.; Huang, W.L.; Qian, H. Identification of highly potent and orally available free fatty acid receptor 1 agonists bearing isoxazole scaffold. Bioorganic Med. Chem. 2018, 26, 703–711.
- Fettach, S.; Thari, F.Z.; Hafidi, Z.; Karrouchi, K.; Bouathmany, K.; Cherrah, Y.; El Achouri, M.; Benbacer, L.; El Mzibri, M.; Sefrioui, H.; et al. Biological, toxicological and molecular docking evaluations of isoxazoline-thiazolidine-2,4-dione analogues as new class of anti-hyperglycemic agents. J. Biomol. Struct. Dyn. 2021, 12, 1072–1084.
- Sysak, A.; Obminska-Mrukowicz, B. Isoxazole ring as a useful scaffold in a search for new therapeutic agents. Eur. J. Med. Chem. 2017, 137, 292–309.
- Tugrak, M.; Gul, H.I.; Bandow, K.; Sakagami, H.; Gulcin, I.; Ozkay, Y.; Supuran, C.T. Synthesis and biological evaluation of some new mono Mannich bases with piperazines as possible anticancer agents and carbonic anhydrase inhibitors. Bioorganic Chem. 2019, 90, 103095.
- Bhardwaj, S.; Bendi, A.; Singh, L. A Study on Synthesis of Chalcone Derived-5-Membered Isoxazoline and Isoxazole Scaffolds. Curr. Org. Synth. 2022, 19, 643–663.
- Chopra, B.; Dhingra, A.K. Natural products: A lead for drug discovery and development. Phytother. Res. 2021, 35, 4660–4702.
- Yao, H.; Liu, J.K.; Xu, S.T.; Zhu, Z.Y.; Xu, J.Y. The structural modification of natural products for novel drug discovery. Expert Opin. Drug Discov. 2017, 12, 121–140.
- Farooq, S.; Ngaini, Z. Synthesis of Benzalacetophenone-based Isoxazoline and Isoxazole Derivatives. Curr. Org. Chem. 2022, 26, 679–692.
- Wang, H.B.; He, Y.; Jian, M.L.; Fu, X.G.; Cheng, Y.H.; He, Y.J.; Fang, J.; Li, L.; Zhang, D. Breaking the Bottleneck in Anticancer Drug Development: Efficient Utilization of Synthetic Biology. Molecules 2022, 27, 7480.
- Chouiab, K.; Romdhane, A.; Delemasure, S.; Dutartre, P.; Elie, N.; Touboul, D.; Ben Jannet, H.; Hamza, M.A. Regiospecific synthesis, anti-inflammatory and anticancer evaluation of novel 3,5-disubstituted isoxazoles from the natural maslinic and oleanolic acids. Ind. Crops Prod. 2016, 85, 287–299.
- Mallavadhani, U.V.; Vanga, N.R.; Jeengar, M.K.; Naidu, V.G.M. Synthesis of novel ring-A fused hybrids of oleanolic acid with capabilities to arrest cell cycle and induce apoptosis in breast cancer cells. Eur. J. Med. Chem. 2014, 74, 398–404.
- Adachi, H.; Nosaka, C.; Atsumi, S.; Nakae, K.; Umezawa, Y.; Sawa, R.; Kubota, Y.; Nakane, C.; Shibuya, M.; Nishimura, Y. Structure-activity relationships of natural quinone vegfrecine analogs with potent activity against VEGFR-1 and-2 tyrosine kinases. J. Antibiot. 2021, 74, 734–742.
- Aissa, I.; Abdelkafi-Koubaa, Z.; Chouaib, K.; Jalouli, M.; Assel, A.; Romdhane, A.; Harrath, A.H.; Marrakchi, N.; Ben Jannet, H. Glioblastoma-specific anticancer activity of newly synthetized 3,5-disubstituted isoxazole and 1,4-disubstituted triazole-linked tyrosol conjugates. Bioorg. Chem. 2021, 114, 105071.
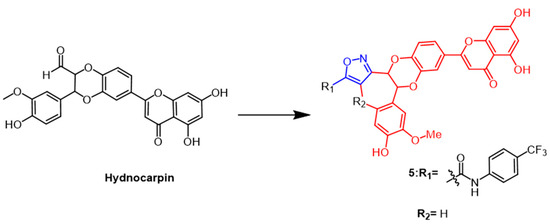
- Mathai, B.M.; Joseph, M.M.; Maniganda, S.; Nair, J.B.; Arya, J.S.; Karunakaran, V.; Radhakrishnan, K.V.; Maiti, K.K. Guanidinium rich dendron-appended hydnocarpin exhibits superior anti-neoplastic effects through caspase mediated apoptosis. RSC Adv. 2016, 6, 52772–52780.
- Arya, J.S.; Joseph, M.M.; Sherin, D.R.; Nair, J.B.; Manojkumar, T.K.; Maiti, K.K. Exploring Mitochondria-Mediated Intrinsic Apoptosis by New Phytochemical Entities: An Explicit Observation of Cytochrome c Dynamics on Lung and Melanoma Cancer Cells. J. Med. Chem. 2019, 62, 8311–8329.
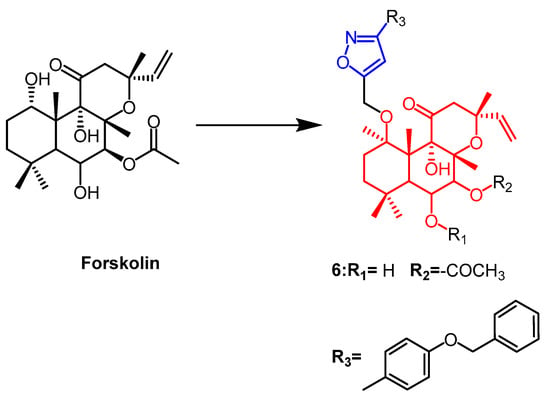
- Burra, S.; Voora, V.; Rao, C.P.; Vijay Kumar, P.; Kancha, R.K.; David Krupadanam, G.L. Synthesis of novel forskolin isoxazole derivatives with potent anti-cancer activity against breast cancer cell lines. Bioorg. Med. Chem. Lett. 2017, 27, 4314–4318.
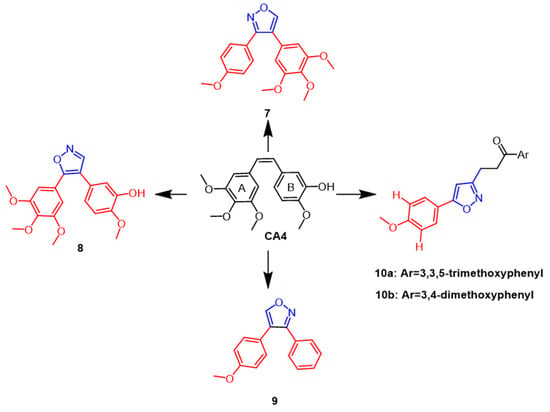
- Chernysheva, N.B.; Maksimenko, A.S.; Andreyanov, F.A.; Kislyi, V.P.; Strelenko, Y.A.; Khrustalev, V.N.; Semenova, M.N.; Semenov, V.V. Regioselective synthesis of 3,4-diaryl-5-unsubstituted isoxazoles, analogues of natural cytostatic combretastatin A4. Eur. J. Med. Chem. 2018, 146, 511–518.
- Semenova, M.N.; Demchuk, D.V.; Tsyganov, D.V.; Chernysheya, N.B.; Samet, A.V.; Silyanova, E.A.; Kislyi, V.P.; Maksimenko, A.S.; Varakutin, A.E.; Konyushkin, L.D.; et al. Sea Urchin Embryo Model As a Reliable in Vivo Phenotypic Screen to Characterize Selective Antimitotic Molecules. Comparative evaluation of Combretapyrazoles, -isoxazoles,-1,2,3-triazoles, and -pyrroles as Tubulin-Binding Agents. ACS Comb. Sci. 2018, 20, 700–721.
- Silyanova, E.A.; Ushkarov, V.I.; Samet, A.V.; Maksimenko, A.S.; Koblov, I.A.; Kislyi, V.P.; Semenova, M.N.; Semenov, V.V. A comparative evaluation of monomethoxy substituted o-diarylazoles as antiproliferative microtubule destabilizing agents. Mendeleev Commun. 2022, 32, 120–122.
- Thiriveedhi, A.; Nadh, R.V.; Srinivasu, N.; Kaushal, K. Novel Hybrid Molecules of Isoxazole Chalcone Derivatives: Synthesis and Study of In Vitro Cytotoxic Activities. Lett. Drug Des. Discov. 2018, 15, 576–582.
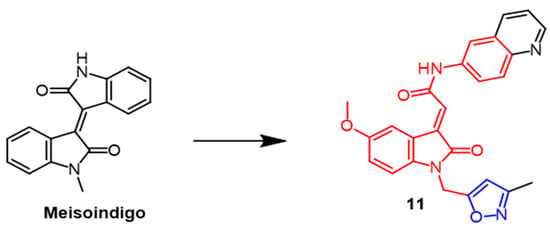
- Chiou, C.-T.; Lee, W.-C.; Liao, J.-H.; Cheng, J.-J.; Lin, L.-C.; Chen, C.-Y.; Song, J.-S.; Wu, M.-H.; Shia, K.-S.; Li, W.-T. Synthesis and evaluation of 3-ylideneoxindole acetamides as potent anticancer agents. Eur. J. Med. Chem. 2015, 98, 1–12.
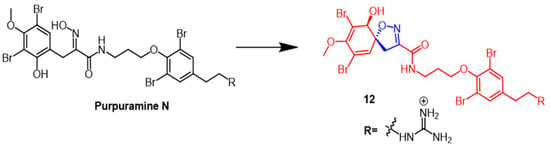
- Dai, J.; Parrish, S.M.; Yoshida, W.Y.; Yip, M.L.R.; Turkson, J.; Kelly, M.; Williams, P. Bromotyrosine-derived metabolites from an Indonesian marine sponge in the family Aplysinellidae (Order Verongiida). Bioorg. Med. Chem. Lett. 2016, 26, 499–504.
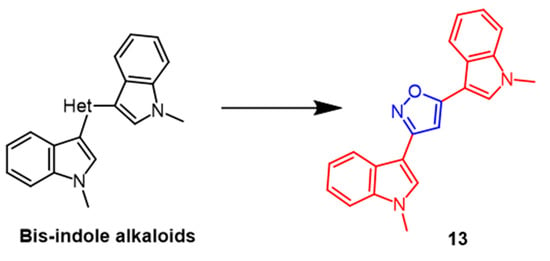
- Diana, P.; Carbone, A.; Barraja, P.; Kelter, G.; Fiebig, H.-H.; Cirrincione, G. Synthesis and antitumor activity of 2,5-bis(3′-indolyl)-furans and 3,5-bis(3′-indolyl)-isoxazoles, nortopsentin analogues. Bioorg. Med. Chem. 2010, 18, 4524–4529.
- Fawzi, M.; Oubella, A.; Bimoussa, A.; Bamou, F.Z.; Khdar, Z.A.; Auhmani, A.; Riahi, A.; Robert, A.; Morjani, H.; Itto, M.Y.A. Design, synthesis, evaluation of new 3-acetylisoxazolines and their hybrid analogous as anticancer agents: In vitro and in silico analysis. Comput. Biol. Chem. 2022, 98, 107666.
- Oubella, A.; Ait Itto, M.Y.; Auhmani, A.; Riahi, A.; Robert, A.; Daran, J.-C.; Morjani, H.; Parish, C.A.; Esseffar, M.H. Diastereoselective synthesis and cytotoxic evaluation of new isoxazoles and pyrazoles with monoterpenic skeleton. J. Mol. Struct. 2019, 1198, 126924.
- Wu, Z.Q.; Wang, C.C.; Huang, M.Z.; Tao, Z.H.; Yan, W.J.; Du, Y.Q. Naturally Occurring Sesquiterpene Lactone-Santonin, Exerts Anticancer Effects in Multi-Drug Resistant Breast Cancer Cells by Inducing Mitochondrial Mediated Apoptosis, Caspase Activation, Cell Cycle Arrest, and by Targeting Ras/Raf/MEK/ERK Signaling Pathway. Med. Sci. Monit. 2019, 25, 3676–3682.
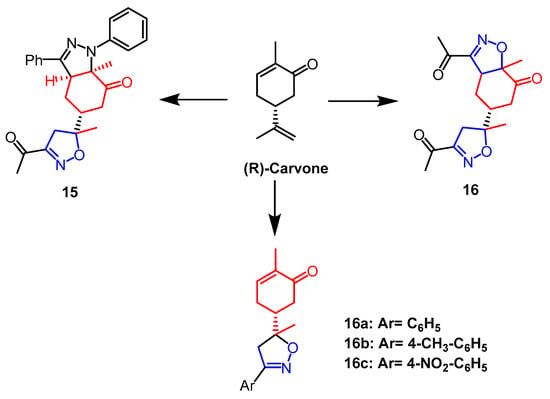
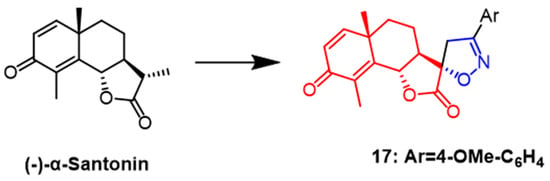
- Khazir, J.; Singh, P.P.; Reddy, D.M.; Hyder, I.; Shafi, S.; Sawant, S.D.; Chashoo, G.; Mahajan, A.; Alam, M.S.; Saxena, A.K.; et al. Synthesis and anticancer activity of novel spiro-isoxazoline and spiro-isoxazolidine derivatives of α-santonin. Eur. J. Med. Chem. 2013, 63, 279–289.
- Ock, C.W.; Kim, G.D. Harmine Hydrochloride Mediates the Induction of G2/M Cell Cycle Arrest in Breast Cancer Cells by Regulating the MAPKs and AKT/FOXO3a Signaling Pathways. Molecules 2021, 26, 6714.
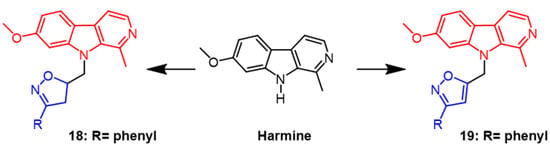
- Filali, I.; Bouajila, J.; Znati, M.; Bousejra-El Garah, F.; Ben Jannet, H. Synthesis of new isoxazoline derivatives from harmine and evaluation of their anti-Alzheimer, anti-cancer and anti-inflammatory activities. J. Enzyme Inhib. Med. Chem. 2015, 30, 371–376.
- Filali, I.; Romdhane, A.; Znati, M.; Jannet, H.B.; Bouajila, J. Synthesis of New Harmine Isoxazoles and Evaluation of their Potential Anti-Alzheimer, Anti-inflammatory, and Anticancer Activities. Med. Chem. 2016, 12, 184–190.
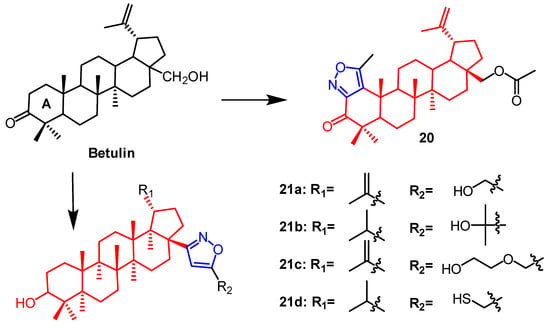
- Luginina, J.; Linden, M.; Bazulis, M.; Kumpins, V.; Mishnev, A.; Popov, S.A.; Golubeva, T.S.; Waldvogel, S.R.; Shults, E.E.; Turks, M. Electrosynthesis of Stable Betulin-Derived Nitrile Oxides and their Application in Synthesis of Cytostatic Lupane-Type Triterpenoid-Isoxazole Conjugates. Eur. J. Org. Chem. 2021, 2021, 2557–2577.
- Ma, L.; Miao, D.Y.; Lee, J.J.; Li, T.; Chen, Y.; Su, G.Y.; Zhao, Y.Q. Synthesis and biological evaluation of heterocyclic ring-fused dammarane-type ginsenoside derivatives as potential anti-tumor agents. Bioorganic Chem. 2021, 116, 105365.
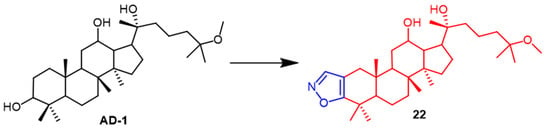
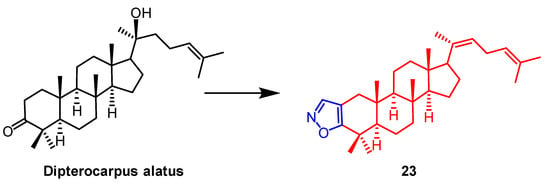
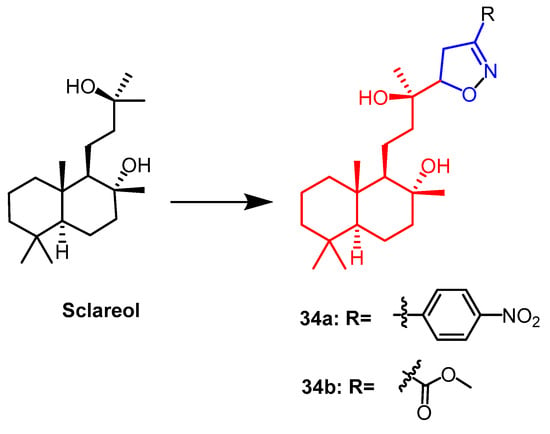
- Smirnova, I.E.; Kazakova, O.B.; Loesche, A.; Hoenke, S.; Csuk, R. Evaluation of cholinesterase inhibitory activity and cytotoxicity of synthetic derivatives of di- and triterpene metabolites from Pinus silvestris and Dipterocarpus alatus resins. Med. Chem. Res. 2020, 29, 1478–1485.
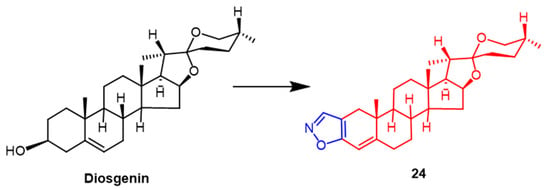
- Erdagi, S.I.; Yildiz, U. Synthesis, Structural Analysis and Antiproliferative Activity of Nitrogen-Containing Hetero Spirostan Derivatives: Oximes, Heterocyclic Ring-Fused and Furostanes. Chemistryselect 2022, 7, e202200439.
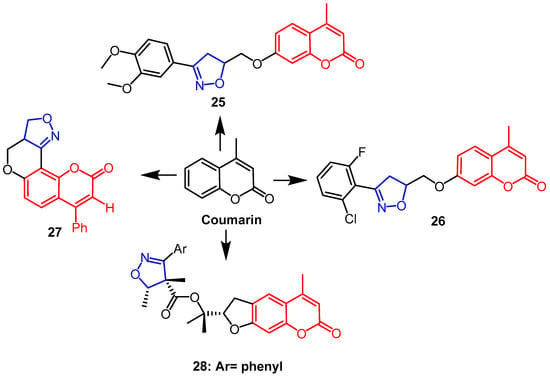
- Lingaraju, G.S.; Balaji, K.S.; Jayarama, S.; Anil, S.M.; Kiran, K.R.; Sadashiva, M.P. Synthesis of new coumarin tethered isoxazolines as potential anticancer agents. Bioorg. Med. Chem. Lett. 2018, 28, 3606–3612.
- Krishna, C.; Bhargavi, M.V.; Rao, Y.J.; Krupadanam, G.L.D. Synthesis of pyrano isoxazoline/isoxazole annulated coumarins via intramolecular nitrile oxide cycloaddition and their cytotoxicity. Russ. J. Gen. Chem. 2017, 87, 1857–1863.
- Znati, M.; Debbabi, M.; Romdhane, A.; Ben Jannet, H.; Bouajila, J. Synthesis of new anticancer and anti-inflammatory isoxazolines and aziridines from the natural (-)-deltoin. J. Pharm. Pharmacol. 2018, 70, 1700–1712.
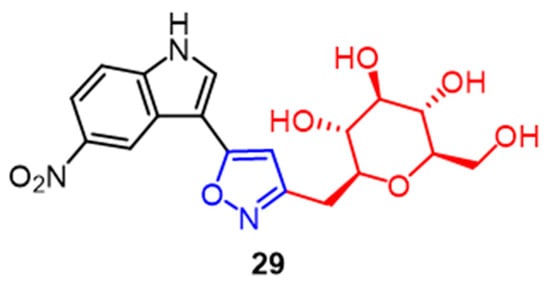
- Kumari, P.; Mishra, V.S.; Narayana, C.; Khanna, A.; Chakrabarty, A.; Sagar, R. Publisher Correction: Design and efficient synthesis of pyrazoline and isoxazole bridged indole C-glycoside hybrids as potential anticancer agents. Sci. Rep. 2020, 10, 10095.
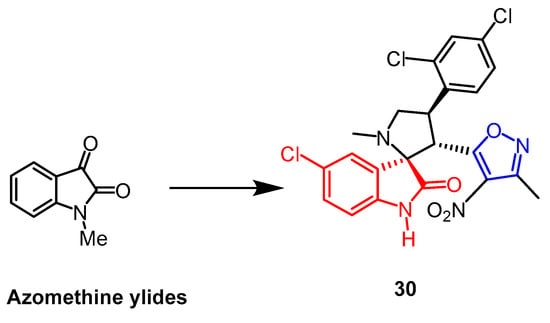
- Liu, X.-W.; Yao, Z.; Yang, J.; Chen, Z.-Y.; Liu, X.-L.; Zhao, Z.; Lu, Y.; Zhou, Y.; Cao, Y. 1,3-Dipolar cycloaddition enabled isoxazole-fused spiropyrrolidine oxindoles syntheses from 3-methyl-4-nitro-5-alkenyl-isoxazoles and azomethine ylides. Tetrahedron 2016, 72, 1364–1374.
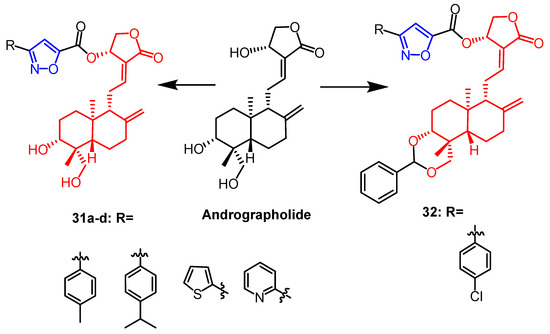
- Mokenapelli, S.; Yerrabelli, J.R.; Das, N.; Roy, P.; Chitneni, P.R. Synthesis and cytotoxicity of novel 14α-O-(andrographolide-3-subsitutedisoxazole-5-carboxylate) derivatives. Nat. Prod. Res. 2021, 35, 3738–3744.
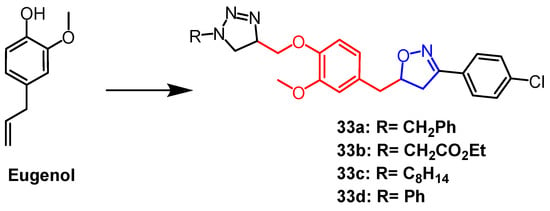
- Oubella, A.; Taia, A.; Byadi, S.; Ait Lahcen, M.; Bimoussa, A.; Essaber, M.; Podlipnik, C.; Morjani, H.; Ait Itto, M.Y.; Aatif, A. Chemical profiling, cytotoxic activities through apoptosis induction in human fibrosarcoma and carcinoma cells, and molecular docking of some 1,2,3-triazole-isoxazoline hybrids using the eugenol as a precursors. J. Biomol. Struct. Dyn. 2022, 40, 1–13.
- Phanumartwiwath, A.; Kesornpun, C.; Sureram, S.; Hongmanee, P.; Pungpo, P.; Kamsri, P.; Punkvang, A.; Eurtivong, C.; Kittakoop, P.; Ruchirawat, S. Antitubercular and antibacterial activities of isoxazolines derived from natural products: Isoxazolines as inhibitors of Mycobacterium tuberculosis InhA. J. Chem. Res. 2021, 45, 1003–1015.
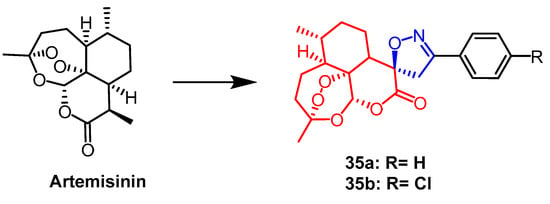
- Pratap, S.; Naaz, F.; Reddy, S.; Jha, K.K.; Sharma, K.; Sahal, D.; Akhter, M.; Nayakanti, D.; Kumar, H.M.S.; Kumari, V.; et al. Anti-proliferative and anti-malarial activities of spiroisoxazoline analogues of artemisinin. Arch. Pharm. 2019, 352, 1800192.
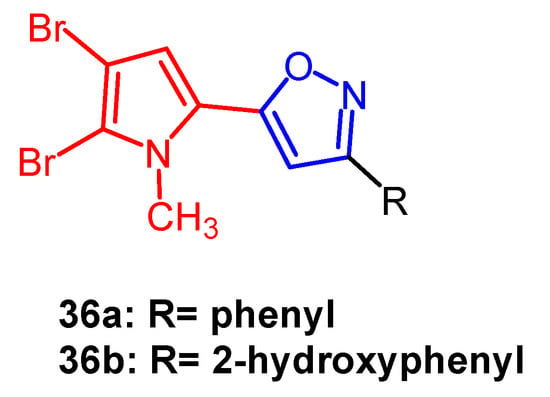
- Rane, R.A.; Sahu, N.U.; Gutte, S.D.; Mahajan, A.A.; Shah, C.P.; Bangalore, P. Synthesis and evaluation of novel marine bromopyrrole alkaloid-based hybrids as anticancer agents. Eur. J. Med. Chem. 2013, 63, 793–799.
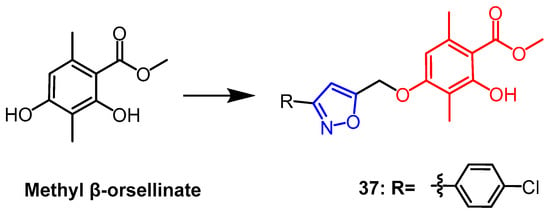
- Reddy, S.T.; Mendonza, J.J.; Makani, V.K.K.; Bhadra, M.P.; Uppuluri, V.M. Synthesis of some novel methyl β-orsellinate based 3, 5-disubstituted isoxazoles and their anti-proliferative activity: Identification of potent leads active against MCF-7 breast cancer cell. Bioorg. Chem. 2020, 105, 104374.
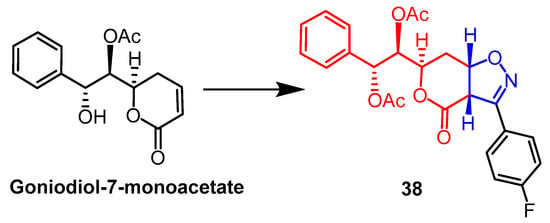
- Talimarada, D.; Sharma, A.; Wakhradkar, M.G.; Dhuri, S.N.; Gunturu, K.C.; Sundaram, V.N.N.; Holla, H. Synthesis, DFT analysis and in-vitro anti-cancer study of novel fused bicyclic pyranone isoxazoline derivatives of Goniodiol-diacetate-a natural product derivative. Fitoterapia 2022, 163, 105316.
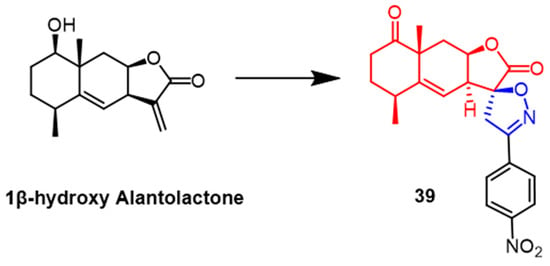
- Tang, J.-J.; He, Q.-R.; Dong, S.; Guo, X.; Wang, Y.-G.; Lei, B.-L.; Tian, J.-M.; Gao, J.-M. Diversity Modification and Structure-Activity Relationships of Two Natural Products 1β-hydroxy Alantolactone and Ivangustin as Potent Cytotoxic Agents. Sci. Rep. 2018, 8, 1722.
- Rodrigues, F.C.; Kumar, N.V.A.; Hari, G.; Pai, K.S.R.; Thakur, G. The inhibitory potency of isoxazole-curcumin analogue for the management of breast cancer: A comparative in vitro and molecular modeling investigation. Chem. Pap. 2021, 75, 5995–6008.
More