Bone is a mineralized tissue composed of calcium phosphates and organic materials such as collagen and proteoglycans. There are two phases of bone mineralization: primary and secondary. Primary mineralization is achieved by osteoblasts. Osteoblasts also produce a large amount of matrix vesicles, which mineralize in nodules (globular assemblies of hydroxyapatite crystals) and then extend into the collagen fibrils secreted by the osteoblasts. In contrast to primary mineralization, secondary mineralization is the process whereby the mineral density of bone increases after primary mineralization. It is postulated that secondary mineralization is regulated through physical crystal maturation, and by the cellular activities of osteocytes embedded in the bone matrix.
- matrix vesicle
- osteoblast
- bone
1. Introduction
2. Cartilage Mineralization by Hypertrophic Chondrocytes
3. Vascular Invasion at the Chondro-Osseous Junction
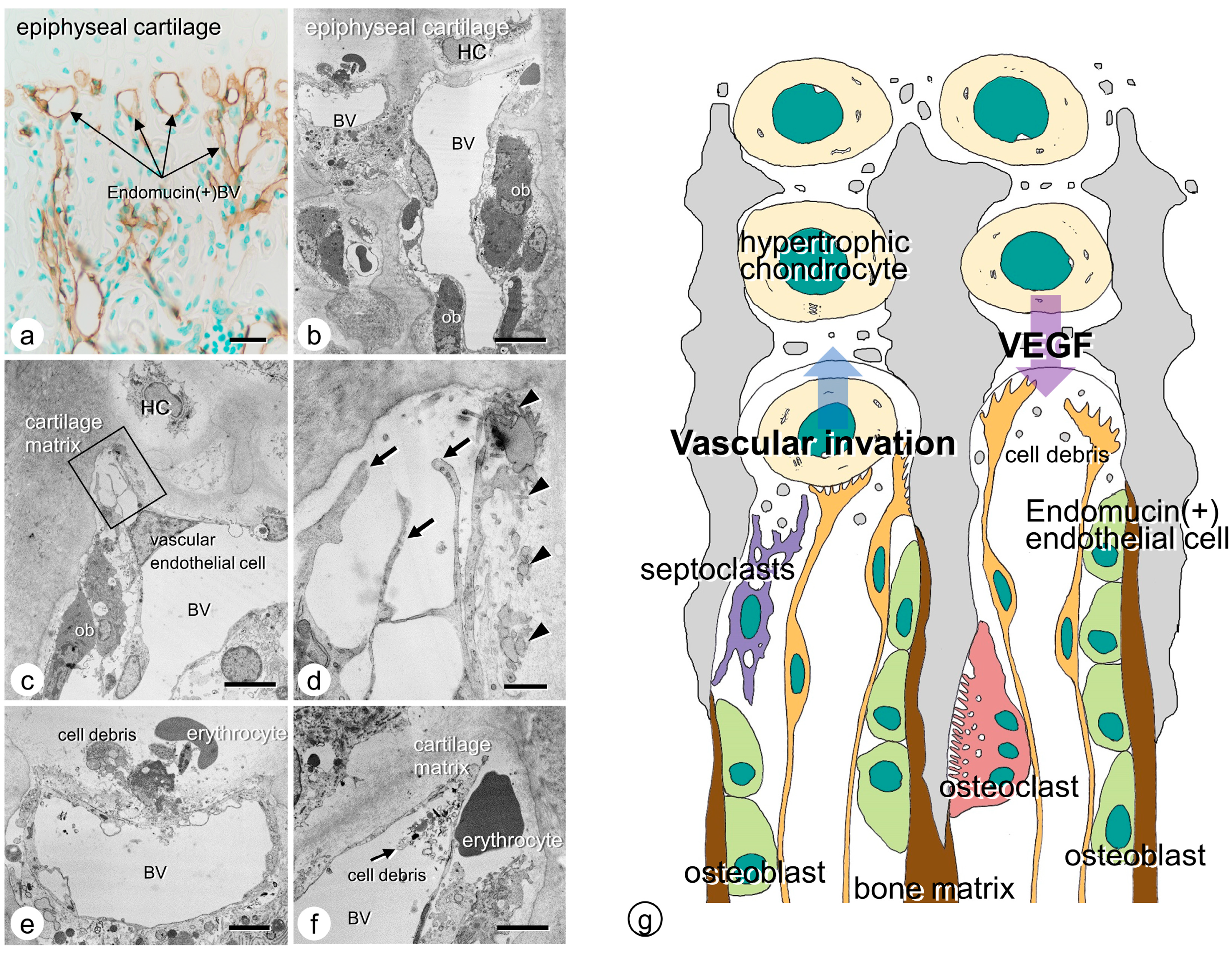
4. Osteoclasts’ Function at the Chondro-Osseous Junction
References
- Amizuka, N.; Hasegawa, T.; Oda, K.; Freitas, P.H.L.; Hoshi, K.; Li, M.; Ozawa, H. Histology of epiphyseal cartilage calcification and endochondral ossification. Front. Biosci. 2012, 4, 2085–2100.
- Ali, S.Y.; Sajdera, S.W.; Anderson, H.C. Isolation and characterization of calcifying matrix vesicles from epiphyseal cartilage. Proc. Natl. Acad. Sci. USA 1970, 67, 1513–1520.
- Anderson, H.C. Vesicles associated with calcification in the matrix of epiphyseal cartilage. J. Cell Biol. 1969, 41, 59–72.
- Bonucci, E. Fine structure of early cartilage calcification. J. Ultrastruct. Res. 1967, 20, 33–50.
- Bonucci, E. Fine structure and histochemistry of “calcifying globules” in epiphyseal cartilage. Z. Zellforsch. Mikrosk. Anat. 1970, 103, 192–217.
- Ozawa, H.; Yamada, M.; Yajima, T. The ultrastructural and cytochemical aspects of matrix vesicles and calcification processes. In Formation and Calcification of Hard Tissues; Talmage, R.V., Ozawa, H., Eds.; Shakai Hoken Pub: Tokyo, Japan, 1978; pp. 9–57.
- Ozawa, H.; Yamada, M.; Yamamoto, T. Ultrastructural observations on the location of lead and calcium in the mineralizing dentine of rat incisor. In Matrix Vesicles; Ascenzi, A., Bonucci, E., de Bernard, B., Eds.; Wiching Editore srl: Milano, Italy, 1981; pp. 179–187.
- Wuthier, R.E. Lipid composition of isolated epiphyseal cartilage cells, membranes and matrix vesicles. Biochim. Biophys. Acta 1975, 409, 128–143.
- Ohba, S. Hedgehog Signaling in Skeletal Development: Roles of Indian Hedgehog and the Mode of Its Action. Int. J. Mol. Sci. 2020, 21, 6665.
- Tavormina, P.L.; Shiang, R.; Thompson, L.M.; Zhu, Y.Z.; Wilkin, D.J.; Lachman, R.S.; Wilcox, W.R.; Rimoin, D.L.; Cohn, D.H.; Wasmuth, J.J. Thanatophoric dysplasia (types I and II) caused by distinct mutations in fibroblast growth factor receptor 3. Nat. Genet. 1995, 9, 321–328.
- Su, W.C.; Kitagawa, M.; Xue, N.; Xie, B.; Garofalo, S.; Cho, J.; Deng, C.; Horton, W.A.; Fu, X.Y. Activation of Stat1 by mutant fibroblast growth-factor receptor in thanatophoric dysplasia type II dwarfism. Nature 1997, 386, 288–292.
- Peters, K.; Ornitz, D.; Werner, S.; Williams, L. Unique expression pattern of the FGF receptor 3 gene during mouse organogenesis. Dev. Biol. 1993, 155, 423–430.
- Deng, C.; Wynshaw-Boris, A.; Zhou, F.; Kuo, A.; Leder, P. Fibroblast growth factor receptor 3 is a negative regulator of bone growth. Cell 1996, 84, 911–921.
- Amizuka, N.; Davidson, D.; Liu, H.; Valverde-Franco, G.; Chai, S.; Maeda, T.; Ozawa, H.; Hammond, V.; Ornitz, D.M.; Goltzman, D.; et al. Signalling by fibroblast growth factor receptor 3 and parathyroid hormone-related peptide coordinate cartilage and bone development. Bone 2004, 34, 13–25.
- Greenspan, J.S.; Blackwood, H.J. Histochemical studies of chondrocyte function in the cartilage of the mandibular codyle of the rat. J. Anat. 1966, 100, 615–626.
- Ikeda, T.; Nomura, S.; Yamaguchi, A.; Suda, T.; Yoshiki, S. In situ hybridization of bone matrix proteins in undecalcified adult rat bone sections. J. Histochem. Cytochem. 1992, 40, 1079–1088.
- Oshima, O.; Leboy, P.S.; McDonald, S.A.; Tuan, R.S.; Shapiro, I.M. Developmental expression of genes in chick growth cartilage detected by in situ hybridization. Calcif. Tissue Int. 1989, 45, 182–192.
- Poole, A.R.; Pidoux, I.; Rosenberg, L. Role of proteoglycans in endochondral ossification: Immunofluorescent localization of link protein and proteoglycan monomer in bovine fetal epiphyseal growth plate. J. Cell Biol. 1982, 92, 249–260.
- Schmid, T.M.; Linsenmayer, T.F. Immunohistochemical localization of short chain cartilage collagen (type X) in avian tissues. J. Cell Biol. 1985, 100, 598–605.
- Gerber, H.P.; Vu, T.H.; Ryan, A.M.; Kowalski, J.; Werb, Z.; Ferrara, N. VEGF couples hypertrophic cartilage remodeling, ossification and angiogenesis during endochondral bone formation. Nat. Med. 1999, 5, 623–628.
- Tsuchiya, E.; Hasegawa, T.; Hongo, H.; Yamamoto, T.; Abe, M.; Yoshida, T.; Zhao, S.; Tsuboi, K.; Udagawa, N.; Freitas, P.H.L.; et al. Histochemical assessment on the cellular interplay of vascular endothelial cells and septoclasts during endochondral ossification in mice. Microscopy 2021, 70, 201–214.
- Kojima, T.; Hasegawa, T.; Freitas, P.H.L.; Yamamoto, T.; Sasaki, M.; Horiuchi, K.; Hongo, H.; Yamada, T.; Sakagami, N.; Saito, N.; et al. Histochemical aspects of the vascular invasion at the erosion zone of the epiphyseal cartilage in MMP-9-deficient mice. Biomed. Res. 2013, 34, 119–128.
- Vu, T.H.; Shipley, J.M.; Bergers, G.; Berger, J.E.; Helms, J.A.; Hanahan, D.; Shapiro, S.D.; Senior, R.M.; Werb, Z. MMP-9/gelatinase B is a key regulator of growth plate angiogenesis and apoptosis of hypertrophic chondrocytes. Cell 1998, 93, 411–422.
- Engsig, M.T.; Chen, Q.J.; Vu, T.H.; Pedersen, A.C.; Therkidsen, B.; Lund, L.R.; Henriksen, K.; Lenhard, T.; Foged, N.T.; Werb, Z.; et al. Matrix metalloproteinase 9 and vascular endothelial growth factor are essential for osteoclast recruitment into developing long bones. J. Cell Biol. 2000, 151, 879–889.
- Marks, S.C., Jr.; Odgren, P.R. The structure and development of the skeleton. In Principles of Bone Biology; Bilezikian, J.P., Raisz, L.G., Rodan, G.A., Eds.; Academic Press: New York, NY, USA, 2002; pp. 3–15.
- Nakamura, H.; Ozawa, H. Ultrastructural, enzyme-, lectin, and immunohistochemical studies of the erosion zone in rat tibiae. J. Bone Miner. Res. 1996, 11, 1158–1164.
- Lee, E.R.; Lamplugh, L.; Shepard, N.L.; Mort, J.S. The septoclast, a cathepsin B-rich cell involved in the resorption of growth plate cartilage. J. Histochem. Cytochem. 1995, 43, 525–536.
- Gartland, A.; Mason-Savas, A.; Yang, M.; MacKay, C.A.; Birnbaum, M.J.; Odgren, P.R. Septoclast deficiency accompanies postnatal growth plate chondrodysplasia in the toothless (tl) osteopetrotic, colony-stimulating factor-1 (CSF-1)-deficient rat and is partially responsive to CSF-1 injections. Am. J. Pathol. 2009, 175, 2668–2675.
- Bando, Y.; Yamamoto, M.; Sakiyama, K.; Inoue, K.; Takizawa, S.; Owada, Y.; Iseki, S.; Kondo, H.; Amano, O. Expression of epidermal fatty acid binding protein (E-FABP) in septoclasts in the growth plate cartilage of mice. J. Mol. Histol. 2014, 45, 507–518.
- Bando, Y.; Yamamoto, M.; Sakiyama, K.; Sakashita, H.; Taira, F.; Miyake, G.; Iseki, S.; Owada, Y.; Amano, O. Retinoic acid regulates cell-shape and -death of E-FABP (FABP5)-immunoreactive septoclasts in the growth plate cartilage of mice. Histochem. Cell Biol. 2017, 148, 229–238.