2. Antitumour Activity
Malignant tumors are extremely complex, and they can have a major impact on people’s life and health. The incidence of cancer has been on the rise, and despite the availability of many drugs and treatments for cancer, it remains one of the greatest threats to human health. Natural products or their derivatives make up over 65% of all anti-cancer medications. Natural products thus serve a crucial clinical role in the treatment of cancer. As biosynthetic technology has advanced, A growing number of natural products are being developed for cancer therapy as clinical candidates
[25]. As a result, scientists have created several isoxazole and isoxazoline derivatives with anti-cancer properties based on natural products.
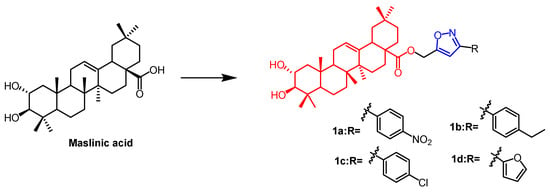
Maslinic acid (MA)(
Figure 1) and oleanolic acid (OA) (
Figure 2) can be isolated from the natural
Olea europaea L. MA as well as OA have been found to have anti-cancer and anti-inflammatory properties. A number of isoxazole-containing pentacyclic triterpene derivatives were created and examined by Chouaïb and coworkers. The majority of the isoxazoles, especially those generated from MA, showed remarkable anti-cancer activity in tests on the cancer cell lines EMT-6 and SW480. Isoxazole derivatives of MA
1a,
1b,
1c, and
1d (
Figure 1) showed better anti-cancer properties against SW480 cell line compared to the starting substrate MA (viability (%/control) = 9, 9, 10, and 10%, respectively, 30 µM). However, only compounds
1d and
1c showed higher activity than MA against EMT6 (breast) (viability (%/control) = 5 and 64%, respectively, 10 µM). The scientists also performed an anti-proliferative assessment on the cancer cell lines EMT-6 and SW480 and documented the synthesis of OA isoxazole derivatives. However, according to in vitro cytotoxicity testing, OA contains a more potent anti-proliferative active compound than its isoxazole derivatives
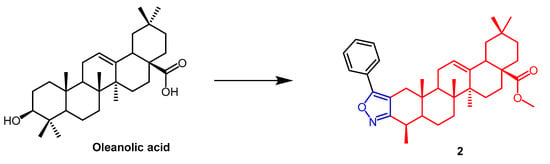
[26]. In another study, A number of OA nitrogen heterocyclic derivatives with nitrogen heterocycles at C-2 and C-3 were created by Mallavadhani et al. According to the study, pyrimidine derivatives had much greater activity than isoxazoles derivative
2 (
Figure 2) against seven cancerous cell lines. The pyrimidine derivatives stopped the cell cycle and caused apoptosis in MCF cells during the S phase, according to the flow cytometric study
[27].
Figure 1. The chemical structure and derivative of maslinic. (The red marker in the figure indicates the parent structure of MA, and the blue marker indicates the structural modification of isoxazole.)
[26].
Figure 2. The chemical structure and derivatives of oleanolic. (The red marker in the figure indicates the parent structure of OA, and the blue marker indicates the structural modification of isoxazole.) The same explanation for the following figures
[27].
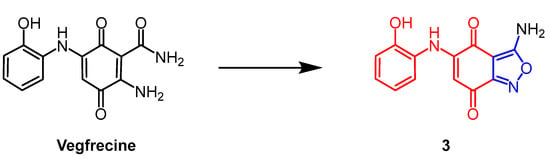
Streptomyces sp. extract vegfrecine (
Figure 3) as a VEGF receptor tyrosine kinase inhibitor. It exhibits potent in vitro inhibitory activity against VEGFR-1 and VEGFR-2 tyrosine kinases by blocking VEGFR-1 signaling which inhibits pathological angiogenesis associated with cancer and tumor metastasis. Adachi et al. synthesized a natural quinone compound containing an isoxazole ring through the structure of vegfrecine. Compound
3 (
Figure 3), with a quinone ring thickened with an isoxazole ring, exhibited moderate inhibitory activity against VEGFR-1 tyrosine kinase (IC
50 = 0.65 µM) and reduced inhibitory activity against VEGFR-2 tyrosine kinase compared to vegfrecine (IC
50 = 7.1 µM). It is explained by the structure–activity relationship (SAR) that the isoxazole ring formed by the aminocarbonyl and amino groups which inhibits VEGFR-1 and VEGFR-2 tyrosine kinases by fixing the orientation of the aminocarbonyl and amino groups
[28].
Figure 3.
The chemical structure and derivatives of vegfrecine [28].
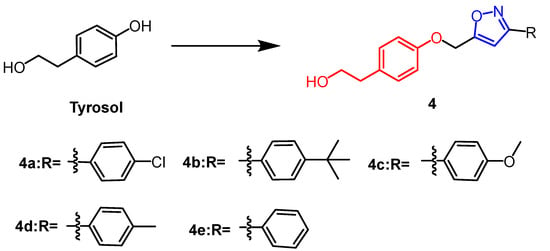
Tyrosol (
Figure 4) is a natural phenolic compound obtained from various plants. In the study by Aissa et al., 3,5-disubstituted isoxazole derivatives (
4a–e) (
Figure 4) were synthesized from tyrosol, of which compounds
4c,
4b, and
4a showed the greatest antiproliferative properties with IC
50 values of 67.6 µM, 42.8 µM, and 61.4 µM, respectively. The derivative
4c was superior to the positive drug than temozolomide (IC
50 = 53.85 µM). The newly synthesized compounds exerted anticancer activity by inducing apoptosis in U87 cells. It was found that methyl, methoxy, or chloride substitutions on the R group of isoxazole derivatives enhanced their activity against U87 cells based on SAR studies
[29].
Figure 4.
The chemical structure and derivatives of tyrosol [29].
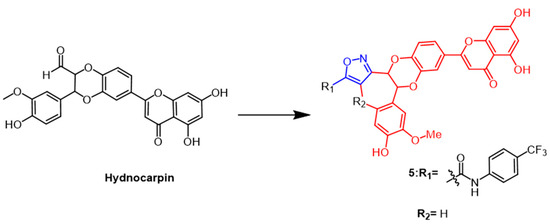
A flavonoid derived from the seeds of
Hydnocarpus wightiana Blume is called hydnocarpin (Hy) (
Figure 5). It was reported that Hy exhibit antitumor effects
[30]. Arya et al. modified Hy as the structural basis by introducing isoxazole rings to develop a series of new compounds. The new derivatives induce apoptosis and arrest the cell cycle at G2/M and S phases in human metastatic melanoma (A375) and human lung adenocarcinoma (A549) cells. One of the most potent compounds was compound
5 (
Figure 5), which inhibited A375 at IC
50 values of 3.6 and 0.76 µM at 24 h and 48 h, respectively, about 18–60 fold higher than Hy. In conclusion, the results indicate that attaching isoxazole to naturally occurring hydnocarpin enhanced the selectivity and cytotoxicity of the derivatives against A549 and A375 cells
[31].
Figure 5.
The chemical structure and derivatives of hydnocarpin [31].
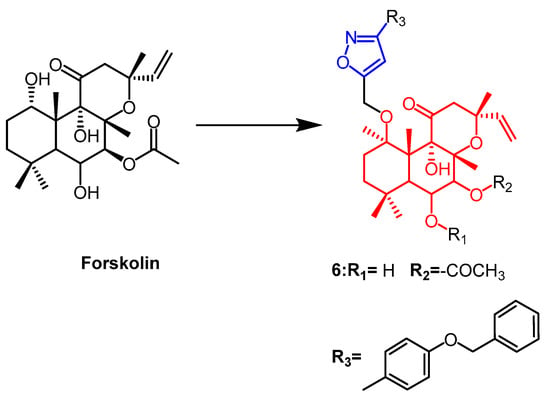
Forskolin (
Figure 6) is a natural product of the labdane diterpene. Its antiproliferative activity was shown to be mediated by the tumor suppressor protein p53. Therefore Burra et al. introduced new isoxazoles by 1,3-dipole cycloaddition reactions at C1-OH of forskolin and tested their activity against breast cancer cell lines. The parental forskolin was active against MCF-7 cells with an IC
50 of 63.3 µM, but did not exhibit any anticancer activity against BT-474 cells (IC
50 > 100 µM). Among the derivatives tested in this study, the compound
6 (
Figure 6) with an acetyl group at the 7th position displayed the highest activity against MCF-7 and BT-474 cell lines, exhibiting an IC
50 of 0.5 µM
[32].
Figure 6.
The chemical structure and derivatives of forskolin [32].
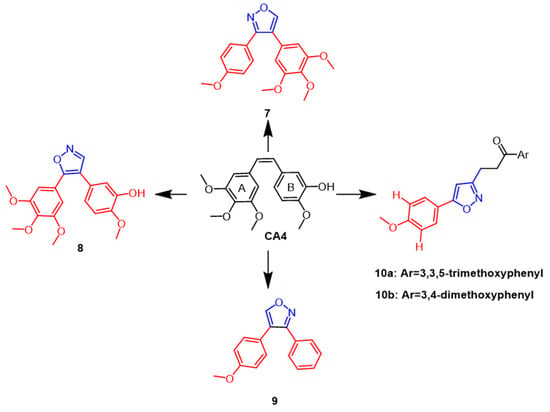
A combretastatin A-4 (CA4) (
Figure 7) isolated from the bark of the African willow tree
Combretum caffrum is being developed as a potent natural cytostatic agent that blocks and apoptoses cancer cells in the G2/M phase. According to SAR studies, the
cis double bonds bound to 3,4,5-trimethoxyphenyl ring A as well as the 4-methoxyphenyl ring B are essential to CA4’s anti-mitotic microtubule destabilizing activity. Using in vivo sea urchin embryo experiments to examine the isoxazole ring modification to CA4, Chernysheva et al. came to the conclusion that CA4 showed anti-mitotic micro-tubule destabilization at a minimum effective concentration (MEC) of 0.002 µM. Chernysheva et al. modified CA4 by introducing isoxazole rings and evaluated them using in vivo sea urchin embryo assays and concluded that CA4 exhibited anti mitotic microtubule destabilization at a minimum effective concentration (MEC) of 0.002 µM, while isoxazole derivatives
7 and
9 (
Figure 7) caused altered division in sea urchin embryos at 0.005 µM and 0.02 µM, respectively
[33]. In the same year, a new series of CA4 derivatives, including diaryl pyrazoles, isoxazoles, and pyrroles, were reported in the literature. To evaluate the antimitotic efficacy of their drugs against microtubules, a panel of human cancer cells and in vivo sea urchin embryo experiments were performed. The strongest antimitotic agent among the isoxazole derivatives was discovered to be compound
8 (
Figure 7) (EC = 0.001 µM), possessing better antiproliferative activity than CA4 (EC = 0.002 µM) while also exhibiting comparable cytotoxicity against human cancer cells. According to structure–activity relationship studies, in the 4,5-diarylisoxazole series, removing the 3-hydroxyl group from ring B decreases the antimitotic activity, and the 3-hydroxyl group is required for antiproliferative activity
[34]. Silyanova and colleagues found that only the isoxazole heterocycle and the unsubstituted benzene ring next to the heteroatom conferred the appropriate conformation of the molecule to exert antiproliferative effects through the microtubule destabilization mode of action. The 4,5-diarylisoxazoles showed greater antimitotic activity than 3,4-diarylisoxazoles
[35]. In 2018, isoxazole chalcone derivatives with structural similarities to CA4 were synthesized. According to the results, the new synthesized compounds
10a and
10b (
Figure 7) exhibited potent cytotoxic activity against DU145 prostate cancer cell lines with IC
50 values of 0.96 µM and 1.06 µM, respectively, compared to the positive control (IC
50 =4.10 µM). According to structure–activity relationship studies, electron-giving on the benzene ring groups, such as methoxy substituents, enhanced the anticancer activity
[36].
Figure 7. The chemical structure and derivatives of CA4 [35][36]. The chemical structure and derivatives of CA4 [35,36].
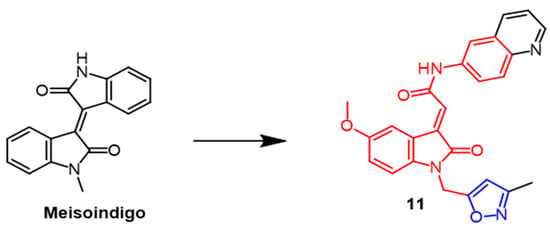
Indirubin is an active ingredient in Chinese medicine formulas with good anti-cancer properties. Meisoindigo (
Figure 8) is derived from Indirubin and is effective against cancer. Therefore, Tang et al. synthesized a series of 3-subunit indoleacetamides using meisoindigo as a structural template. Different cancer cell lines were tested by researchers to see if they were cytotoxic. Such compounds arrest the cell cycle in the G1 phase and subsequently trigger cystatinase-dependent apoptosis. Compound
11 (
Figure 8) showed the best activity in the series with IC
50 values of 2.3, 2.7, 2.2, 3.6, and 3.6 µM against MCF-7, Hep3B, KB, SF-268, and MKN-48 cancer cell lines, respectively. In comparison with meisoindigo, compound
11 had better antiproliferative properties against these five cancer cell lines (IC
50 = 25.5, 9.0, 19.2, 37.0, 36.7 µM). As a result of adding isoxazole to precursor compounds, their antiproliferative properties were enhanced
[37].
Figure 8.
The chemical structure and derivatives of meisoindigo [37].
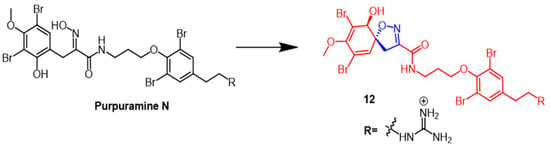
Dai et al. identified many novel bromotyrosine-derived compounds from the
Indonesian sponge. The isolated compound purpuramine N (
Figure 9) was modified by oxidation of aromatic groups to produce derivative
12 (
Figure 9) containing an isoxazoline fraction. Unfortunately, NIH3T3 cells (normal mouse fibroblasts) were inhibited by compound
12, so this compound was not further investigated. However, further studies could be conducted with compound
12 due to its moderate inhibition of aspartate protease BACE1 (memapsin-2)
[38].
Figure 9.
The chemical structure and derivatives of pupuramine N [38].
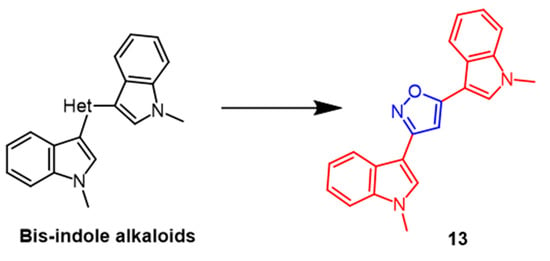
There are several biological activities associated with
bis-indole alkaloids, which are sponge metabolites. Several novel
bis-indolyl-isoxazoles and
bis-indolyl-furans were synthesized and evaluated as antitumor agents in 10 human tumor cell lines, according to Diana et al. The most potent compound among the isoxazole derivatives was
13 (
Figure 10), displaying mean IC
50 values of 53.2 µM. Compound
13 was detected to have selective activity against A549 and LXFA 629L (lung) and UXF 1138L (uterine body). The results indicated that the bisindolyl isofuran derivatives exhibited more significant antitumor activity than isoxazole derivatives against human tumor cell lines
[39].
Figure 10.
The chemical structure and derivatives of
bis
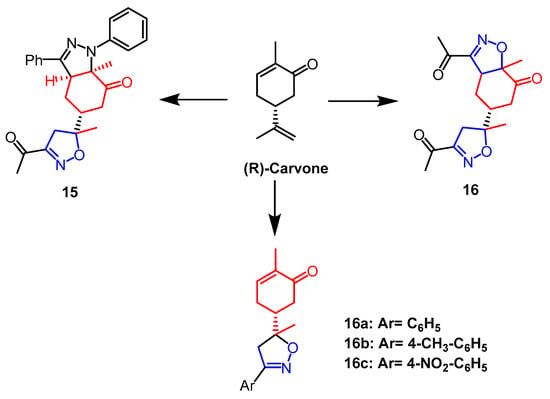
(R)-Carvone belongs to a group of monoterpenes that are present in many natural products and bioactive molecules. An array of derivatives of monoterpenes was synthesized by Fawzi et al. Through MTT assays, all synthesized molecules were evaluated for cytotoxicity against HT-1080, A-549, MCF-7, and MDA-MB-231 cells, which concluded that compound
15 (
Figure 11), an isoxazole-pyrazole heterodimer, had no significant activity against all selected cancer cell lines without significant activity. In terms of growth inhibition, compound
14 (
Figure 11) was the strongest with IC
50 values of 22.47, 25.87, 19.19, and 20.79 µM, respectively. Analyses of the SAR process revealed that the two isoxazoline parts of compound
14 are responsible for the cytotoxicity of the compound on human cancer cells. Further studies by flow cytometry showed that compound
14 caused MCF-7 cancer cells and MDA-MB-231 cancer cells to arrest in the S and G2/M phases of the cell cycle, as well as induced early apoptosis of MCF-7 and MDA-MB-231 through caspase-3/7 activation
[40]. In research by Oubella and colleagues, chiral isoxazolines and pyrazole derivatives with monoterpene backbones were efficiently synthesized from (R)-Carvone. Human HT1080, MCF-7, and A-549 cancer cells were used as test subjects for the newly synthesized monoterpene isoxazoline and pyrazole derivatives’ cytotoxic properties. Among them, isoxazoline derivatives
16a,
16b, and
16c (
Figure 11) showed the best anticancer activity against HT1080 cells with IC
50 values of 16.1 µM, 10.72 µM, and 9.02 µM, respectively. In contrast, pyrazole derivatives were less active in HT1080 cells, all with IC
50 values over 100 µM. In HT-1080 cells, isoxazoline derivatives exhibited a greater anticancer activity than pyrazole derivatives
[41].
Figure 11. The chemical structure and derivatives of (R) -carvone [40][41]. The chemical structure and derivatives of (R) -carvone [40,41].
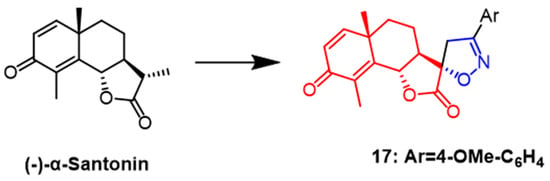
(–)-
α-Santonin is a sesquiterpene lactone compound derived from various Asian plants. Recently, it was shown by flow cytometry that naturally occurring santonin can cause G2/M phase arrest of SK-BR-3 cancer cells in the cell cycle while inhibiting the expression of cell cycle proteins A and B1 and also exerts anticancer effects by blocking the Raf/MEK/ERK pathway in breast cancer cells
[42]. In a recent study, Khazir et al. synthesized new spirocyclic derivatives of the human santonin and tested their anticancer activity against cancer cell lines Among them, spiroisoxazoline derivative
17 (
Figure 12) showed good activity against MCF-7 and A549 cell lines with IC
50 values of 0.02 and 0.2 µM, respectively. As a result, compound
17 is promising as a new anticancer drug
[43].
Figure 12.
The chemical structure and derivatives of (–)-
α
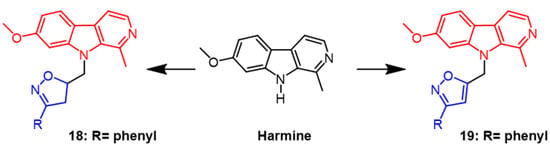
Harmine can be extracted from natural
Peganum harmala seeds. According to research, it has significant cytotoxic activity against cancer cell lines and can induce the G2/M cell cycle arrest in breast cancer cells by regulating MAPK and AKT/FOXO3a signaling pathways
[44]. Harmine was used as a scaffold to generate derivatives containing isoxazoline, and its cytotoxicity against MCF7 breast cancer and HCT116 colon cancer was assessed. As a result of the tests, Harbin had potent cytotoxicity on both cells with IC
50 values of 0.7 µM and 1.3 µM, respectively. Among the synthesized derivatives, derivative
18 (
Figure 13) with a benzene ring in the isoxazoline part showed the best activity with IC
50 values of 9.7 µM and 0.2 µM, respectively. According to the report, the cytotoxic activity of the derivative isoxazoline part decreases when the aromatic system of the derivative bears’ methyl, methoxy, and Cl atoms in the para position, respectively
[45]. Next, the group synthesized isoxazole derivatives from harmine and evaluated ovarian cancer (OCVAR-3), breast cancer (MCF-7), and colon cancer (HCT 116) cell lines using MTT assays. Among the synthesized derivatives, compound
19 (
Figure 13) showed the best activity with IC
50 values of 5.0, 16.0, and 5.0 μM, respectively. It can be seen that the isoxazoline derivatives showed more bioactivity in the colon cancer (HCT 116) cell line
[46].
Figure 13. The chemical structure and derivatives of phenyl [45][46]. The chemical structure and derivatives of phenyl [45,46].
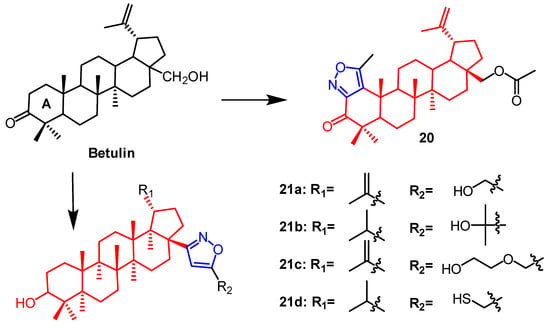
Plants contain betulin, which is a pentacyclic triterpene naturally occurring in many species. Lugiņina et al. synthesized dense heterocyclic derivatives based on this natural compound. The isoxazole ring was joined to the betulin scaffold by altering the triterpene ring A. Using the MTT assay, the cytotoxic activity of each derivative was evaluated against the human cancer cell lines RD TE32, A549, MS, HEp-2, and HCT 116. Among them, the N-acetyl triazole of betulin showed the strongest activity with IC
50 2.3–7.5 µM, whereas the thickened isoxazole ring derivative
20 (
Figure 14) was not as active as the triazole derivatives with IC
50 values of 7.9–22.1 µM. The synthetic and cytotoxic activity of betulin derivatives containing the isoxazole fraction was reported by Lugiņina et al. Five tumor cell lines were used to test the effectiveness of all derivatives (A-549, MDA-MB-231, MCF-7, KB, and KB-VIN). Compound
21a (
Figure 14) showed GI
50 values of 11.05 ± 0.88 µM against the lung cancer cell line A549. Compounds
21b and
21c (
Figure 14) were found against breast cancer MCF7 cells (11.47 ± 0.84 µM) and (14.51 ± 1.42 µM). Compound
21d (
Figure 14) was found on cells of breast cancer MCF7 (12.49 ± 1.18 µM) and lung cancer A549 (13.15 ± 1.56 µM). The constitutive relationship indicated that compounds with hydrophilic substituents on the isoxazole ring had stronger cytotoxic activity
[47].
Figure 14.
The chemical structure and derivatives of betulin [47].
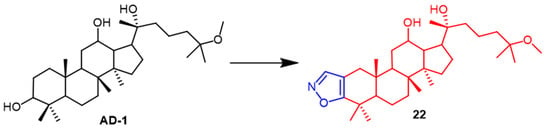
AD-1 (
Figure 15) is a novel ginsenoside discovered to induce G0/G1 cell cycle arrest, apoptosis, and ROS production. Ma et al. introduced various heterocycles containing nitro groups at the C-2 and C-3 positions to synthesize AD-1 derivatives. The AD-1 IC
50 value of 14.38 μM for isoxazole derivative
22 (
Figure 15) showed solid cellular activity, which enriched our approach to studying the synthesis of AD-1 derivatives
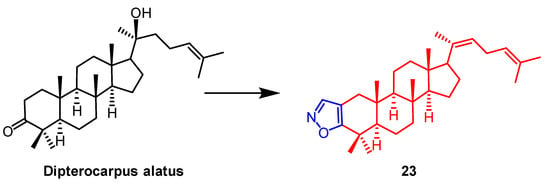
[48]. Smirnova et al. synthesized a
Dipterocarpus alatus derivative
23 containing the isoxazole fraction and evaluated its cholinesterase inhibitory activity and cytotoxicity. Compound
23 (
Figure 16) was found to be cytotoxic to MCF7 (breast cancer) (EC
50 = 16.2 µM) and mildly inhibited the enzyme AChE with 15.9% inhibition. This study provides some reference for the structural modification of
D. alatus [49].
Figure 15.
The chemical structure and derivatives of AD-1 [48].
Figure 16. The chemical structure and derivatives of
Dispterocarous alatus [49].
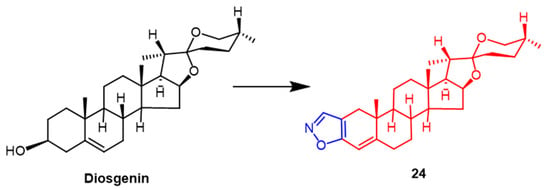
The roots of a few wild yams contain large amounts of diosgenin. Diosgenin has received a lot of attention as a possible anticancer drug in recent years. Yildiz et al. synthetically modified the diosgenin backbone to obtain some new derivatives. The findings demonstrated that among these compounds, The pyridine-containing compound had the highest efficacy against human breast cancer (MCF-7), with an IC
50 value of 5.72 µM. Compound
24 (
Figure 17), containing the isoxazole fraction, also showed potent anticancer activity against human breast cancer (MCF-7) and lung adenocarcinoma (A549) with IC
50 values of 9.15 ± 1.30 µM and 14.92 ± 1.70 µM, which was superior to the parent compound diosgenin (IC
50 = 26.91 ± 1.84 µM and 36.21 ± 2.42 µM). It can be inferred that the A-ring substituted isoxazole and pyrazole fractions may enhance the anticancer activity of diosgenin derivatives
[50].
Figure 17.
The chemical structure and derivatives of diosgenin [50].
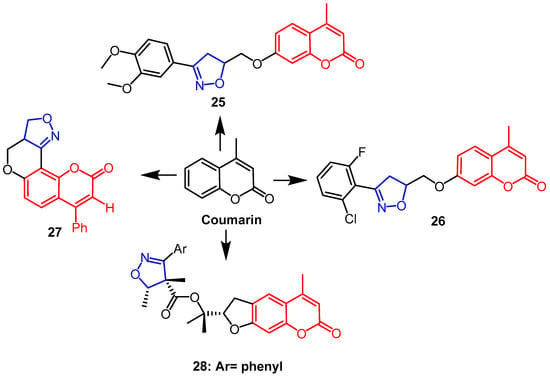
In an in vitro MTS experiment, Lingaraju et al. synthesized coumarin-isoxazoline adducts and assessed their capacity to cause cytotoxicity in human melanoma cancer cell line (UACC 903) and fibroblast normal cell line (FF2441). The findings revealed that compounds
25 and
26 (
Figure 18) exhibited better cytotoxicity against UACC 903, with IC
50 values of 1.5 µM and 4.5 µM, respectively. Compound
26 was the most active, which may be due to the presence of chlorine and fluorine substitutions in the
ortho-positions of the phenyl ring of isoxazoline. Because compound
25 has 3,4-dimethoxy on the benzene ring of the isoxazoline ring, it is more selective for melanoma cancer cells than normal cells, which may explain why compound
25 was the leading contender in this series
[51]. Another team synthesized isoxazoline/isoxazole fused coumarin analogs and evaluated their cytotoxicity against human colorectal cancer (Colo-205), human hepatocellular liver cancer (HepG2) and human cervical cancer (HeLa). The findings revealed that compound
27 (
Figure 18) had more sensitive activity against the HepG2 cell line than Colo-205 and HeLa cell lines, and it had the highest anti-proliferative activity (IC
50 ≤ 50 µM) against all cell lines
[52]. (–)-Deltoin is a coumarin-containing natural product extracted from flowers of
Ferula lutea (Poir.). As potential anticancer agents based on (–)-deltoin, Znati et al. synthesized coumarin derivatives containing isoxazoline backbones. When it came to the human colon cell line HCT-116, compound
28 (
Figure 18) was the most effective (IC
50 = 3.3 µM), four times as effective as the parent chemical (IC
50 = 14.3 µM). This finding suggests that the introduction of the isoxazoline fraction yielded better anticancer potential
[53].
Figure 18. The chemical structure and derivatives of coumarin [51][52][53]. The chemical structure and derivatives of coumarin [51,52,53].
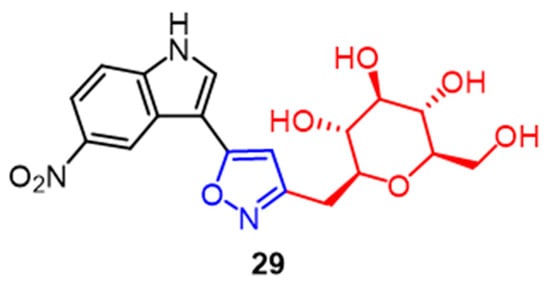
A significant family of compounds known as C-glycosides is present in many natural product architectures and exhibits a wide range of biological activities. Compound
29 (
Figure 19), with an IC
50 value of 0.67 M, showed the greatest cytotoxicity against MCF-7 breast cancer cells among a series of C-glycoside-linked pyrazoline and isoxazole derivatives synthesized by Kumari et al. It can be seen that pyrazoline partially favors the C-glycoside derivatives in COX-2 enzyme inhibition. The addition of the isoxazole ring to the pyrazoline structure further improves the compounds’ biological activity
[54].
Figure 19.
The chemical structure and derivatives of c-glycosides [54].
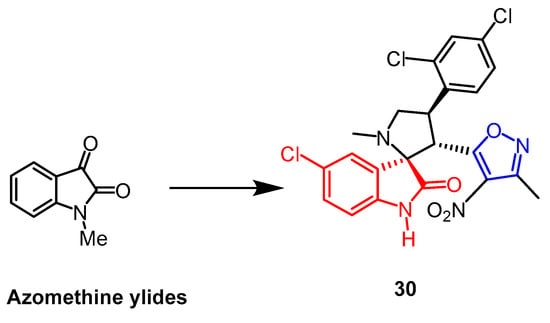
The spiro-pyrrolidine-oxindole ring system has specific structural properties and potent biological activities. A number of isoxazole derivatives of spiroazolidine-oxindole were created by Liu et al. Using an MTT assay, and they assessed the derivatives’ cytotoxic effects on human leukemia cell K562, human prostate cancer cell PC-3, and human lung cancer cell A549. The outcomes demonstrated that compound
30 (
Figure 20) exhibited considerable cytotoxicity against these three cell lines, K562, A549, and PC-3, with IC
50 values of 10.7 µM, 21.5 µM, and 13.1 µM, respectively. Isoxazole was added to the sporozoite–oxindole complex, and it inhibited cancer cell proliferation as well as or better than cisplatin (up to 2.1-fold)
[55].
Figure 20.
The chemical structure and derivatives of azomethine ylides [55].
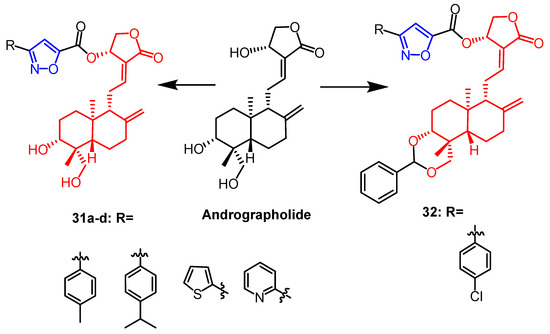
It was reported that a series of isoxazole derivatives were synthesized using naturally occurring andrographolide as a backbone by Mokenapelli et al. The cytotoxicity of the derivatives was also evaluated against HCT15, HeLa, and DU145 cell lines. Compounds
31a,
31b,
31c,
31d, and
32 (
Figure 21) exhibited significant cytotoxicity against the three cancer cell lines with IC
50 values below 40 µg/mL. Therefore, isoxazoline derivatives of andrographolide at the C-14 position are promising as anticancer agents
[56].
Figure 21.
The chemical structure and derivatives of andrographolide [56].
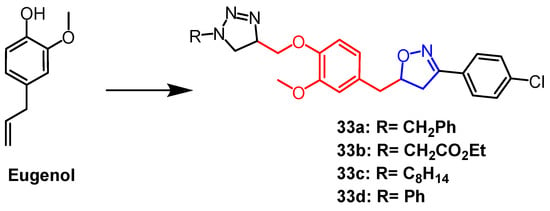
Eugenol is a naturally occurring phenolic monoterpene obtained from clove oil having a variety of biological properties. Using eugenol as a scaffold, Oubella et al. created 1,2,3-triazole mixed isoxazoline derivatives. They next tested and assessed the derivatives’ in vitro anticancer activity against the fibrosarcoma (HT-1080), breast cancer (MCF-7 and MDA-MB-231), and lung cancer (A-549) cell lines. The mixed compounds
33a–d (
Figure 22) exhibited more significant cytotoxicity than the triazole derivatives (IC
50 = 15–29 µg/mL) against the three cancer cell lines. According to preliminary structural investigations, the simultaneous presence of 1,2,3-triazole and isoxazole produced stronger anticancer effects. Follow-up studies showed that the most potent compound
33a, induced apoptosis through the activation of caspase-3/7, leading to cell cycle arrest in A-549 cancer cells in the G2/M phase
[57].
Figure 22.
The chemical structure and derivatives of eugenol [57].
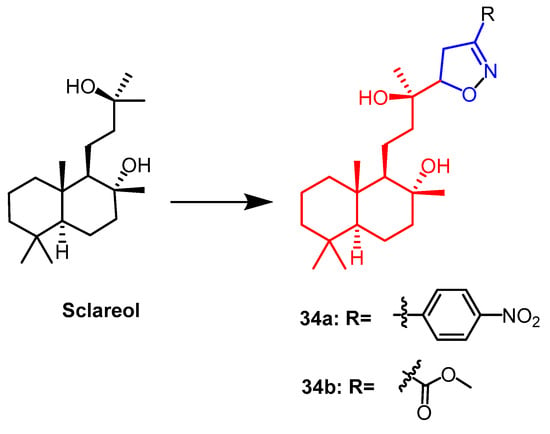
Sclareo is a natural product with anticancer activity. As one of the isoxazoline derivatives of sclareol, derivative
34a (
Figure 23) showed the strongest anticancer activity with cytotoxic activity (IC
50 = 13.20–21.16 µM) against human hepatocellular carcinoma (HepG2), human cholangiocarcinoma (HuCCA-1) and human lung adenocarcinoma (A549) cell lines. When compared to the natural parent chemical sclareol, the findings demonstrated that derivative
34a increased cytotoxicity against cancer cell lines (IC
50 = 49.89–70.40 µM). This could be due to the structure of the derivative showing a significant hydrophobicity (due to the aliphatic skeleton) and hydrogen bonding ability (–OH), leading to increased cellular uptake of the compound, resulting in cytotoxicity
[58].
Figure 23.
The chemical structure and derivatives of sclareol [58].
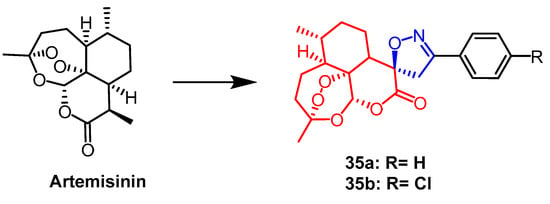
Pratap et al. synthesized a series of artemisinin derivatives containing spiroisoxazoline. They evaluated the antiproliferative activity of the newly synthesized derivatives against the human lung cancer cell line (A-549), human colon cancer cell line (HCT-15), and human liver cancer cell line (Hep-G2) by MTT assay. Among all compounds, compound
35a (
Figure 24) was found to show significant cytotoxicity against all selected cell lines with IC
50 values of 32.43, 4.04, and 46.30 µM, respectively. it can be seen that compound
35a was more sensitive against colon cancer cell line (HCT-15). Compound
35a containing the spiroisoxazoline fraction (IC
50 = 4.04 µM) was nine times more active against HCT-15 cell line than the positive drug 5-fluorouracil (IC
50 = 35.53 µM). Follow-up DNA cell cycle analysis showed that
35a inhibited cell proliferation in the G2/M phase. It was also found that compound
35b (
Figure 24) had significant activity against
P. falciparum (IC
50 = 0.1 µM)
[59].
Figure 24.
The chemical structure and derivatives of artemisinin [59].
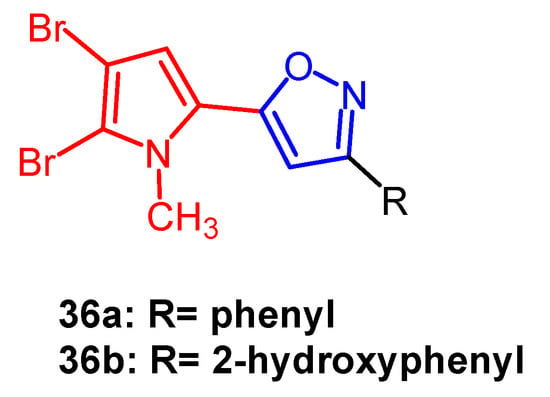
Bromopyrrole alkaloids are an essential family of marine alkaloids with a wide range of biological activities. Rane et al. synthesized a series of isoxazole-containing bromopyrrolidine alkaloid derivatives and evaluated their in vitro antiproliferative activity against five human cancer cell lines by MTT assay. Among them, compound
36a (
Figure 25) exhibited the most potent anti-cancer activity. It was able to selectively inhibit oral cancer cell line KB403 with an IC
50 of 2.45 µM, whereas compound
36b (
Figure 25) (IC
50 = 16.58 µM) was found to be selectively cytotoxic against colon cancer cells CaCO2. Therefore, introducing brominated pyrroles into isoxazoles could enhance the anticancer activity of such compounds
[60].
Figure 25.
The chemical structure and derivatives of bromopyrrole [60].
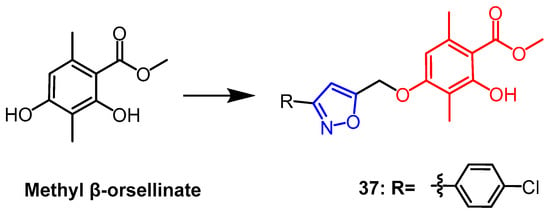
Methyl
β-orsellinate is a highly functionalized natural phenolic molecule with a variety of biological properties that is present in plants. A series of isoxazole derivatives of methyl
β-orsellinate were synthesized by Reddy et al., and four human cancer cell lines as well as the normal cell line HEK-293T (embryonic kidney) were evaluated for their antiproliferative activity in vitro. The majority of these artificial compounds demonstrated antiproliferative efficacy. The most effective combination was found to be compound
37 (
Figure 26), whose IC
50 against the MCF-7 breast cancer cell line was found to be 5 times higher (IC
50 = 7.9 ± 0.07 µM) than the parent compound (IC
50 = 46.63 ± 0.11 µM). The constitutive relationship analysis indicated that chlorine substitution at the benzene ring para position gave the compound a better anticancer potential. Compound
37 was shown by flow cytometric analysis to induce apoptosis and arrest the cell cycle in the G2/M phase
[61].
Figure 26.
The chemical structure and derivatives of methyl
β
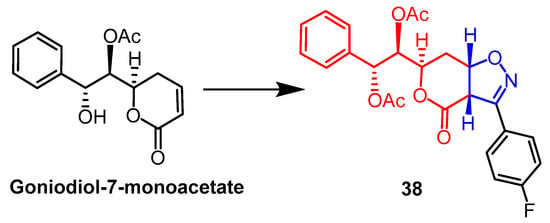
The mono-acetate of goniodiol-7-monoacetate was obtained from ethyl acetate extract of
Goniothalamus wynaadensis Bedd. Goniodiol diacetate was transformed into a new isoxazoline derivative by Talimarada et al. The MTT test was used to measure the derivatives’ cytotoxic activity against the human cancer cell lines MDA-MB-231, SKOV3, PC-3, and HCT-15, as well as the normal human cell line HEK 293. All isoxazoline derivatives were inhibiting cancer cells (EC
50 < 10 µM) without damaging normal cell lines, with compound
38 (
Figure 27) showing the strongest activity, compared to the positive control drug vincristine (EC
50 = 9.02 µM, 7.00 µM), and compound
38 (EC
50 = 6.83 µM, 6.88 µM) on SKOV3 and MDA-MB-231 cell lines exhibited better cytotoxicity. The data suggest that the presence of saturated lactones is essential for activity and that changes in the electron-absorbing or electron-donating groups on the aryl rings of all derivatives have little effect on enhancing cytotoxicity. Further molecular biology studies showed compound
38 stalled the cell cycle in the S phase
[62].
Figure 27.
The chemical structure and derivatives of goniodiol-7-monoacetate [62].
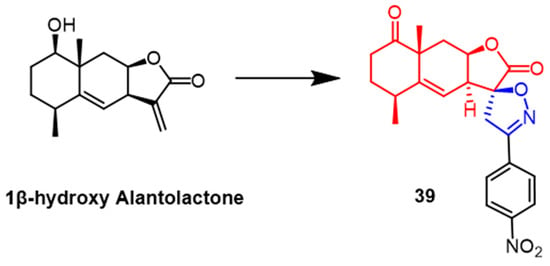
A number of spiroisoxazoline derivatives based on the natural substance 1-hydroxy alantolactone have recently been created by Tang et al. Among them, compound
39 (
Figure 28) exhibited the most potent antitumor activity with IC
50 values of 2.7-5.1 µM against HeLa, PC-3, HEp-2, and HepG2 cells, respectively, which were superior to the parent compound 1β-hydroxy alantolactone (IC
50 = 3.2–6.4 µM). Preliminary conformational analysis indicated that oxidation of the lead compound C1-OH would show greater cytotoxicity; the double bond at the C5–C6 position might be more effective for activity. At the same time, the C1-OH esterified derivative would be less potent. Further studies revealed that compound
39 would concentration-dependently inhibit TNF-
α-induced NF-
κB signaling in PC-3 cancer cells and lead to G2/M phase arrest of PC-3 cancer cells in the cell cycle
[63].
Figure 28.
The chemical structure and derivatives of 1
β
-hydroxy alantolactone [63].
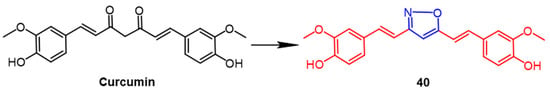
Curcumin is an extremely potent natural product with numerous biological effects. Researchers improved the stability of the compounds by using heterocyclic substitution of the diketone group of curcumin. One of the isoxazole curcumin derivatives,
40 (
Figure 29), exhibited potent antitumor activity, with compound
40 (IC
50 = 3.97 µM) showing more significant cytotoxicity against the breast cancer cell line (MCF7) compared to the parent compound curcumin (IC
50 = 21.89 µM). In addition, compound
40 consistently showed better docking fractions than the other compounds and curcumin. As indicated by preliminary conformational analyses, curcumin’s biological activity was enhanced by the introduction of the isoxazole ring, and isoxazole curcumin is promising as an anti-breast cancer drug
[64].
Figure 29.
The chemical structure and derivatives of curcumin [64].