Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Qikai Qin and Version 3 by Dean Liu.
Glucagon-like peptide-1 receptor (GLP-1R) is a critical therapeutic target for type 2 diabetes mellitus (T2DM). The GLP-1R cellular signaling mechanism relevant to insulin secretion and blood glucose regulation has been extensively studied. Numerous drugs targeting GLP-1R have entered clinical treatment. NHowever, novel functional molecules with reduced side effects and enhanced therapeutic efficacy are still in high demand.
- GLP-1R signaling
- functional molecules
- incretin therapy
1. The Structural Basis of GLP-1R
G protein-coupled receptors (GPCRs) are widely distributed in various tissues and play key roles in a diversity of physiological activities. As the largest receptor family, GPCRs are important drug targets for a broad range of indications [1][20]. GPCRs share a conserved seven-transmembrane helix bundle (Figure 1) with three extracellular loops (ECLs) and three intracellular loops (ICLs). The ECLs form an extracellular surface that interacts with orthosteric ligands. While the ICLs, to large extent, determine downstream receptor signaling. GLP-1R, together with four other glucagon receptors (GCGR, GLP-2R, GIPR, and GHRHR), belongs to the secretin (class B1) GPCR family, whose endogenous ligands are peptide hormones [2][3][21,22]. Class B1 GPCRs have a large and structurally conserved extracellular domain (ECD) of 120–160 residues at the N-terminal, forming a three-layered α-β-β-α fold that is stabilized by three interlayer disulfide bonds [4][23].
The endogenous ligands of GLP-1R are GLP-1 (7-36) and GLP-1 (7-37), products from the post-translational processing of proglucagon [5][24]. Proglucagon also produces several other peptide hormones for receptors in the glucagon receptor family, such as glucagon, oxyntomodulin (OXM), and GLP-2. On binding with endogenous peptides, the glucagon receptor family shares a similar recognition mode, which is described as a “two-domain” binding mode. The C-terminal α helix of peptide ligand initiates peptide recognition by binding to the ECD, then the peptide N-terminal can activate the receptor and trigger its downstream signaling cascade by binding to the transmembrane domain (TMD) ligand-binding pocket [6][25]. Recently released cryo-EM structures of GLP-1R in complex with a peptide ligand revealed that peptides form a single helix in binding post, which is a unique feature shared in class B1 GPCRs [7][8][9][26,27,28].
2. Signaling Pathways of GLP-1R
Researchers have been pursuing functional studies of GLP-1R for many years to illuminate the mechanism of GLP-1R signaling. GLP-1R downstream signaling pathways network can be activated through coupling with the intracellular transducers [10][29]. The diverse protein-binding forms will lead to complex downstream pathways.
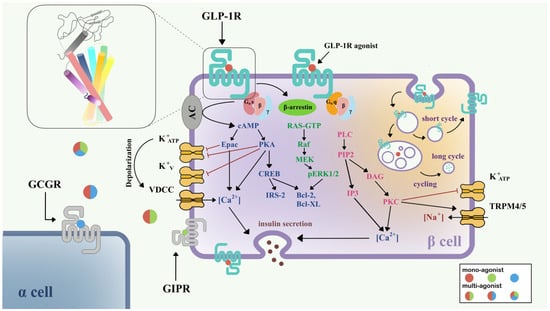
Figure 1. Signaling pathways of GLP-1R in pancreatic β-cell [11][12][15,30]. Three downstream signaling pathways initiated from Gαs (blue), Gαq (pink), and β-arrestin (green) are shown in the left half of the β-cell. The GLP-1R internalization process and insulin secretion after GLP-1R activation are also shown. Colored circles show mono-agonists targeting GLP-1R (red), GIPR (green), and GCGR (blue), and multi-agonists targeting GLP-1R/GIPR (red/green), GLP-1R/GCGR (red/blue), and GLP-1R/GIPR/GCGR (red/green/blue). The top left inset shows the structure of GLP-1R with a seven-transmembrane helix bundle and a large ECD. Abbreviations: VDCC, voltage-dependent calcium channel; TRPM, the transient receptor potential melastatin; GCGR, glucagon receptor; GIPR, gastric inhibitory polypeptide receptor.
Currently, it is believed that GLP-1R predominantly signals through the Gαs/cAMP pathway; however, there is evidence that GLP-1R couples with Gαq and other G proteins. After activation, GLP-1R undergoes phosphorylation at the C-terminal, which further recruits β-arrestin, leading to internalization and desensitization of the receptor (Figure 1). The Gαs/cAMP pathway directly leads to the glucose-induced secretion of insulin granules [13][14][31,32]. After activation by full agonists such as GLP-1, GLP-1R couples with Gαs, activates adenylate cyclase (AC), and causes the accumulation of cAMP [15][33]. With increasing cAMP levels, protein kinase A (PKA) [13][31] and the exchange protein directly activated by cAMP-2 (Epac-2) [16][34] are also activated. PKA and Epac-2 trigger the closure of KATP and KV channels, which depolarizes the cell membrane, opens voltage-dependent calcium channels (VDCC), and causes Ca2+ influx [17][18][35,36]. In addition to the classical function of cAMP, the cAMP/CREB pathway could also induce the expression of insulin receptor substrate 2 (IRS2) and promote β-cell survival, demonstrating the protective effects of GLP-1 analogs on β-cells [19][37].
In addition to the Gαs/cAMP pathway, GLP-1R is also able to couple with other G protein subtypes including Gαi, Gαq, Gαo, and Gα11 [20][21][38,39]. Recent research has demonstrated the importance of the Gαq pathway in the pancreatic β-cell. First, the Gαq pathway initiates from the coupling of phospholipase-C (PLC), transforming phosphatidylinositol 4,5-bisphosphate (PIP2) to inositol triphosphate 3 (IP3) and diacylglycerol (DAG), and accompanies the activation of protein kinase C (PKC) and intracellular Ca2+ influx production via IP3 receptor [21][39]. PKC also triggers the closure of the KATP channel and activation of TRPM4/5 [22][40]. Gαs and Gαq regulate the cAMP and Ca2+ levels, respectively; however, variational mechanisms exist in the complex network of G protein signaling pathways. A switch of Gαs and Gαq pathways has been found under certain conditions, including persistent depolarization of cell membrane [12][30], and reduction in GLP-1 concentration to picomolar [23][41]. The switch of G protein pathways clarifies a possible mechanism for basal insulin secretion under drug treatment and provides important direction for incretin therapy in T2DM.
Two types of β-arrestin, β-arrestin-1 and β-arrestin-2, mediate the GLP-1R downstream signaling in β-cells, and also elicit the receptor internalization process. β-arrestin-1 mediates the phosphorylation of CREB and ERK1/2 [24][42], further phosphorylating Bad (Bcl-xL/Bcl-2-associated death promoter homolog) and inhibiting cell apoptosis [25][43]. β-arrestin-2 functions as an indispensable insulin regulator. Knocking out β-arrestin-2 in the mice model and leading to impaired insulin secretion [26][44]. With the treatment of sulfonylureas, direct interaction between β-arrestin-1 and Epac-2 can upregulate the Ca2+ concentration [27][45].
GLP-1R is a fast-internalized and recycling receptor [28][46]. After activation, GLP-1R-ligand complexes enter the endosome [14][32]. A portion of that is eventually transported to lysosomes for degradation, while the other portion returns to the cell membrane [28][46]. Different agonists may show different effects on GLP-1R internalization and recycling. For example, GLP-1 is apt to receptor recycling, while exendin-4 may favor slower recycling and lysosome targeting [29][47]. The latest study suggested that internalized endosomal GPCR-Gαs complexes are the origin of induced ERK activity [30][48], but the detailed mechanism remains implicit.
Due to advantageous physiological effects and rapid response in GLP-1R signaling pathways, functional molecules targeting GLP-1R are attracting increasing attention in drug discovery. Optimization of the current molecules and discovery of novel compounds are highly demanded.
3. Functional Molecules Targeting GLP-1R
3.1. Peptide-Based Mono-Agonists
The short circulation time of endogenous GLP-1R agonists, such as GLP-1, limits its potential for T2DM treatment. Fortunately, exendin-4, a 39-amino acid peptide from Gila monster (Heloderma suspectum) venom, showed similar characteristics to the mammalian incretin hormone [31][32][49,50]. It naturally has better resistance to DPP-4 cleavage due to its different amino acid sequence (Figure 2A), and thus has a much longer circulation time than GLP-1. Eventually, exendin-4 was developed as the first marketed twice-daily GLP-1R peptide agonist, exenatide [33][51]. Although exenatide represents a huge step forward, antidiabetic drugs with even longer circulation times for better patient compliance are still in demand.
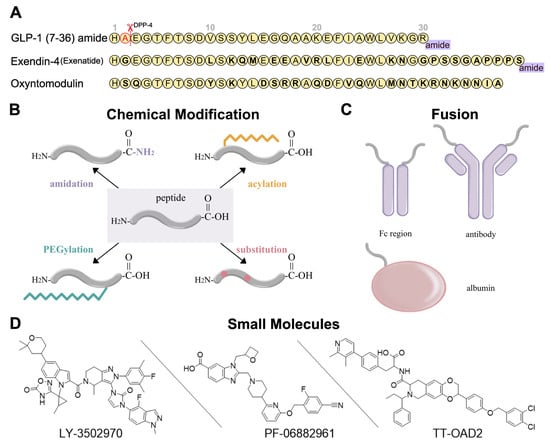
Figure 2. Development strategies for GLP-1R ligands [34][35][36][16,52,53]. (A) Amino acid sequences of GLP-1 (7-36) amide, exendin-4, and oxyntomodulin. Different residues from GLP-1 are in bold type. DPP-4 deactivates the GLP-1 (7-36) amide by cleaving it after the second amino acid (alanine) from the N-terminal and leads to the short circulation time of GLP-1 (7-36) in human body. (B) Several chemical modifications and their combinations can be applied in GLP-1R peptide agonist development, including C-terminal amidation, acylation, PEGylation, and substitution. (C) GLP-1R peptide ligands can fuse to Fc region of antibody, antibody, and albumin. (D) Three typical small-molecule agonists for GLP-1R: LY-3502970 and PF-06882961 are under clinical trials, while TT-OAD2 has failed.
Peptide optimization strategies for GLP-1R peptide agonists include sequence alteration, chemical modification, and fusion. The most obvious approaches to resist degradation are sequence alteration (Figure 2A) and amino acids substitution with α-amino iso-butyric acid (Aib) at the cleavage site [37][38][39][54,55,56] (Figure 2B). As for chemical modification, C-terminal amidation, PEGylation, fatty acid acylation, and amino acid substitution are the main methods to improve peptide stability in vivo (Figure 2B). C-terminal amidation can not only limit proteolytic degradation, thus extending the circulation time of the peptide, but also improve the binding affinity with the receptor [40][57]. PEGylation attaches polyethylene glycol (PEG) units to the peptide [41][58], while fatty acid acylation allows the covalent attachment of a fatty acid chain on the peptide [42][59] (Figure 2B). PEGylation and fatty acid acylation can increase the molecular mass of peptides, improving resistance to renal filtration and enzymatic degradation due to steric hindrance [36][43][53,60]. Beyond that, fatty-acid acylated peptides can spontaneously oligomerize at the subcutaneous injection site and delay absorption due to their amphiphilicity, resulting in a sustained-release effect [44][45][46][61,62,63]. The fatty acid chain can also protect the peptide monomer by reversibly binding to albumin [36][53]. These strategies are usually undertaken in conjunction with each other to generate a better protective effect.
In addition to chemical modifications, reswearchers can also fuse the peptide to a long-circulating and low immunogenic protein, such as an antibody fragment (Fc region), antibody, and albumin (Figure 2C). This strategy can prolong the peptide half-life in two ways. First, like PEGylation and fatty acid acylation, the increased molecular mass can improve resistance to enzymatic degradation and renal filtration. Second, the fusion protein can go through the neonatal Fc receptor (FcRn) recycling pathway [47][64].
3.2. Peptide-Based Multi-Agnosits
Rather than single agonists targeting only GLP-1R, dual or triple agonists targeting additional receptors (GIPR, GCGR, and GLP-2R) involved in the incretin axis and/or other pathways are expected to have stronger glucoregulatory, weight-reducing, and even cardiovascular protective effects [48][4]. Although the mechanism by which multi-agonists have superior effects is unknown, many clinical results have shown their therapeutic potential.
Thus far, GIPR/GLP-1R is the most attractive target combination. Instead of playing a redundant role, GIP may protect β-cells from dysfunction and destruction independently of GLP-1 [49][83]. Although the therapeutic potential of GIPR agonists is debatable, clinical trials of dual GIPR/GLP-1R agonists have produced promising results. The combination of these two GPCR pathways is assumed to increase glucose-dependent insulin secretion, decrease energy consumption, improve white adipose tissue function, and increase insulin sensitivity [50][84]. Tirzepatide, the first dual agonist for the treatment of T2DM and obesity targeting GIPR and GLP-1R, was approved by the U.S. FDA in 2022. Compared with existing drugs such as semaglutide, once-weekly tirzepatide showed better results in glycated hemoglobin A1c (HbA1c) [51][85] and body weight reduction [52][86] with a similar safety profile.
GCGR/GLP-1R dual agonists have also received much attention. OXM can naturally activate both GCGR and GLP-1R with lower potency than their primary ligands [53][87]. Although glucagon and GLP-1 have distinct effects on glucose levels, their effects on food intake may be additive [54][88], leading to more significant body-weight reduction. Several OXM derivatives are under clinical trials, for example, cotadutide, a once-daily GCGR/GLP-1R dual agonist, has shown promising impacts on glycemic control, body weight, and liver fat reduction [55][89].
Clinical trial failures may occur in some cases of dual or triple agonists, caused by their side effects. In order to develop safer therapeutic drugs, it is essential to study the mechanism of these dual and triple agonists [54][88] from structural, functional, and pharmacologic aspects.
3.3. Small Molecule Agonists and Positive Allosteric Modulators (PAM)
So far, all marketed GLP-1R agonists are peptide-based and were developed from natural products such as GLP-1 and exendin-4. Although their half-lives can be prolonged by the strategies discussed above, problems or limits such as cost, side effects, and subcutaneous injection remain [56][100]. In the expectation of improving these deficiencies, many groups and major pharmaceutical companies have long been pursuing the development of non-peptide drugs. Due to a poor understanding of the ligand binding mode and activation mechanism prior to the first GLP-1R structure being solved in 2017 [57][101], high-throughput screening was adapted in many studies to identify promising candidates, followed by massive structure–activity relationship (SAR) studies to improve the chemical and pharmacokinetic properties of compounds.
Currently, none of the small molecule GLP-1R agonists have been approved. However, several candidates are under clinical trial. There are fewer small molecule agonists and PAMs targeting GLP-1R than peptide-based drug candidates, and most of them are still in the early stage of development. One of the first small-molecule agonists, Boc5, is a substituted cyclobutane identified by HT screening [58][107]. However, it has not been launched into clinical study. PF-06882961, based on diazabenzimidazoles, is a full agonist in cAMP elevation, but a partial agonist in other signaling pathways [59][60][108,109]. TTP-273, which has completed phase 2 trials, is an azoanthracene derivative reported in several patents [61][62][110,111]. Another compound in the same series as TTP-273, TT-OAD2, is a partial agonist with slow kinetics in promoting cAMP [63][106]. LY3502970 is a pyrazolopyridine derivative and is a biased agonist that abolished β-arrestin signaling [64][65][103,112]. The chemical structures of PF-07081532 and RGT-075 have not yet been disclosed.
Several GLP-1R structures in complex with different small molecules have been released later, and revealed dramatic diversity in the binding mode of each compound (Figure 3A). TT-OAD2 forms a U-shape conformation in the GLP-1R helical bundle near the ECD where it interacts with residues within the transmembrane helix 1 (TM1) to TM3 and ECLs [63][106]. LY3502970 sits in a similar position as TT-OAD2, while both its arms insert into slits between helixes, clamping around TM2. PF06882961 stays in the orthosteric binding pocket in the TMD, almost overlapping with the binding sites of the N-terminal of GLP-1 [9][28]. Additionally, both PF06882961 and LY3502970 have vital π–π interactions with W33 in the ECD, which abolished their effects on GLP-1R in rodents [64][103]. Finally, a Boc5 binding structure is published recently, which shows that Boc5 adapts a claw shape in its binding pocket, with three fingers stuck into the slits between TM1-TM3 and TM7 [66][113].
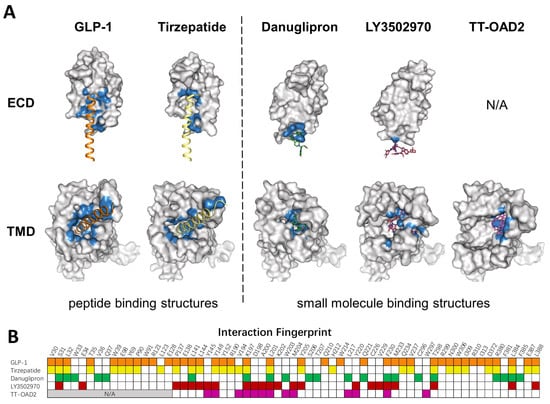
Figure 3. Comparison of peptide and small molecule binding pockets. (A) Structures of GLP1R in complex with GLP1 (PDB: 6X18), tirzepatide (PDB: 7RGP), danuglipron (PDB: 6X1A), LY3502970 (PDB: 7E14), and TT-OAD2 (PDB: 6ORV) are shown in the separated ECD and TMD. GLP-1R is shown as a gray surface model. Residues within 4 Å of the corresponding ligand are colored in blue. (B) Specific residues that interact with corresponding ligands in a fingerprint array. Ligands in (A) and their interaction fingerprints in (B) appear as follows: GLP-1 (orange), tirzepatide (yellow), danuglipron (green), LY3502970 (red), TT-OAD2 (magenta). N/A, not applicable. ECD was not resolved in TT-OAD2 binding structure.
Many allosteric modulators have also been reported in the past few years. As one of the first reported small molecules, compound 2 was extensively studied over the years and was characterized as an ago-PAM [67][68][114,115], a type of molecule that possesses functions of both agonist and PAM. Another set of ago-PAMs based on pyrimidines represented by BETP was identified by screening a small library generated through a pharmacophore model [69][116]. Recently, a new ago-PAM, LSN3318839, was reported to restore the activity of GLP-1 (9-39) [70][117]. However, none of these allosteric modulators has entered clinical study.
To mimic peptide interaction and to obtain sufficient affinity when occupying the peptide pocket, most small molecules end up having a relatively large molecular weight, which could affect the physical properties and pharmacokinetics of compounds. Though most PAMs or ago-PAMs have lower molecular weight, none of them has entered clinical trial. As revealed by the structures, TT-OAD2, LY3502970, and PF-06882961 have quite different binding pockets: only three residues interact with all three compounds, 13 residues interact with two out of three, and 22 residues interact with only one of them (Figure 3B). Different binding modes of each compound may contribute to differences in efficacy and biased agonism. As peptide-based drug development has entered the era of dual agonists, a clearer understanding of the activation and biased signaling mechanism of GLP-1R is needed to aid the design of ideal compounds.