Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by wenyao zhang and Version 2 by Sirius Huang.
Nitrogen–fixing bacteria execute biological nitrogen fixation through nitrogenase, converting inert dinitrogen (N2) in the atmosphere into bioavailable nitrogen. Elaborating the molecular mechanisms of orderly and efficient biological nitrogen fixation and applying them to agricultural production can alleviate the “nitrogen problem”. Azotobacter vinelandii is a well–established model bacterium for studying nitrogen fixation, utilizing nitrogenase encoded by the nif gene cluster to fix nitrogen. In Azotobacter vinelandii, the NifA–NifL system fine–tunes the nif gene cluster transcription by sensing the redox signals and energy status, then modulating nitrogen fixation.
- biological nitrogen fixation
- nitrogenase
- NifA–NifL system
- biological nitrogen fertilizer
1. Introduction
Nitrogen is the core component of biological molecules, such as proteins and nucleic acids. Nitrogen makes up as much as 78% of the atmosphere, but it is not utilized directly by most creatures and can be effectively absorbed only when converted into ammonia or ammonium salts. An increase in available nitrogen content can significantly improve the yield of crops in poor soils [1]. At present, the best way to increase the available nitrogen content of crops is by applying chemical nitrogen fertilizer. However, there are two major disadvantages: First, the utilization rate of nitrogen fertilizer is extremely low, and excessive application of nitrogen fertilizer leads to water eutrophication and causes an imbalance in the species distribution in the water ecosystem, thus resulting in the gradual extinction of the whole water ecosystem. Second, for many smallholders in some developing countries, such as Sub–Saharan Africa, the scarce availability and high cost of nitrogen fertilizer make it unusable; therefore, these smallholders suffer from low yields [2][3][4][5][2,3,4,5]. Finding clean alternatives to nitrogen fertilizers is essential for sustainable and safe agricultural development.
The vast majority of nitrogen fixation is executed by nitrogen–fixing microorganisms [6]. Biological nitrogen fixation is the process in which nitrogen–fixing microorganisms use nitrogenase to directly reduce atmospheric nitrogen to ammonia [7]. This process introducing nitrogen–fixing bacteria or nitrogen–fixing enzymes to the crop provides an opportunity to increase the available nitrogen content of the crop and improve crop nutrition.
Biofertilizers are cheaper, require less capital to use, and are thus increasingly important to agriculture. Biofertilizers, also known as biological inoculants, are organic preparations containing microorganisms [8]. When used as a seed treatment or when seedling roots are immersed in seeds or soil fertilization, they proliferate rapidly and form dense populations in the rhizosphere that rapidly fix nitrogen, increasing the available nitrogen content of the crop. Beneficial biofertilizers for crop production contain nitrogen–fixing bacteria, azospirals, cyanobacteria, green algae, etc. [9][10][11][9,10,11]. However, the application of most nitrogen–fixing microorganisms as biological nitrogen fertilizers in agricultural production faces various obstacles, such as harsh growth conditions and low nitrogen–fixation efficiency. Overcoming these obstacles with biotechnology will facilitate the wide application of biological nitrogen fertilizers in agricultural production, increasing the available nitrogen content of the crop for large agricultural returns.
According to the characteristics of nitrogen fixation, nitrogen–fixation microorganisms can be divided into three types: autogenous nitrogen–fixation bacteria, symbiotic nitrogen–fixation bacteria, and combined nitrogen–fixation bacteria [12]. Autogenous nitrogen–fixing bacteria are free–living bacteria that can fix nitrogen. Azotobacter vinelandii (A. vinelandii), with its high expression and nitrogenase activity, can perform efficient nitrogen fixation under aerobic conditions and is thus becoming a model bacterium for the study of autogenous nitrogen–fixing bacteria [13]. A. vinelandii contains three types of nitrogenases, all of which are multi–subunit protein complexes that bind metal ions, namely, ferric–molybdenum (Fe–Mo) nitrogenase, ferric–vanadium (Fe–V) nitrogenase, and ferric–ferric (Fe–Fe) nitrogenase [14]. The nitrogen–fixing reactions are mainly catalyzed by Fe–Mo nitrogenase, whose expression is regulated by a NifA–NifL system.
2. Nitrogenase and Its Transcriptional Regulation
2.1. Nitrogenase
Nitrogenase is a complex oxygen–sensitive metalloenzyme with three isoforms: Fe–Mo nitrogenase (encoded by nif gene cluster), Fe–V nitrogenase (encoded by vnf gene cluster), and Fe–Fe nitrogenase (encoded by anf gene cluster) (Figure 1a). All known diazotrophs contain at least one of the three closely related nitrogenase isoforms. Although they have different metal contents, these nitrogenase isoforms are related to each other in terms of their structure, mechanism of action, and phylogeny [15].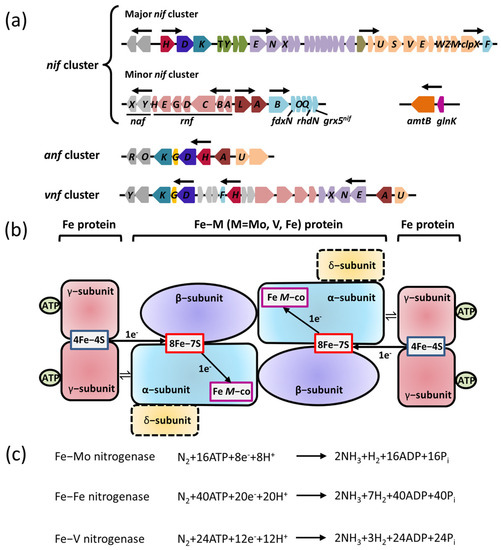
Figure 1. Genes encoding the three forms of nitrogenases and their regulatory proteins are required for nitrogen fixation. (a) The organizations of nif, anf, and vnf clusters encoding the three forms of nitrogenases in A. vinelandii. Predicted σ54–dependent promoter regions are depicted by arrows. Shown are genes encoding the regulatory proteins with known or predicted functions (dark brown), the components involved in the catalytic reduction of N2 (nifD in blue; nifK in aqua; nifH in wine red; nifG in yellow), the assembly or stability of nitrogenase (orange), the maturation of nitrogenase (olive), the maturation of Fe–M cofactor (purple), electron transfer (light red), the biosynthesis of Fe–M cofactor (light aqua), and an unknown function (gray). (b) A diagram of the three forms of nitrogenases involved in electron transfer. M is Mo, Fe, or V. The α–subunit is encoded by nifK; β–subunit is encoded by nifK; γ–subunit is encoded by nifH; δ–subunit is encoded by nifG. (c) The nitrogen–fixation reaction catalyzed by the three forms of nitrogenases.
2.2. Transcriptional Regulation of Nitrogenase
Nitrogenase is extremely sensitive to oxygen, and its catalytic reduction of nitrogen molecules to ammonia must be carried out in a strictly anaerobic microenvironment. As result, the expression of nitrogenase requires a specific microenvironment, where strict anaerobic activity is necessary to maintain the activity of nitrogenase. Additionally, the cell should be in the peak metabolic stage with a large amount of ATP and sufficient nitrogen to ensure the precise modulation of nitrogenase expression by nitrogen–fixing bacteria. The biosynthesis of active nitrogenases relies on a variety of Nif proteins encoded by nif genes beyond the structural subunits of the catalytic center, including the molecular scaffold protein gene, the metal cluster carrier protein gene, and the metal cofactor biosynthesis gene. It was reported that at least nine nif genes are required for the synthesis of bioactive Mo–Fe nitrogenases: nifH, nifE, nifN, nifS, nifU, nifV, nifY, nifB and nifQ, which performs functions including redox provisioning and electron transport [20]. The transcriptional regulation of nitrogenase might differ between nitrogen–fixing bacteria. For instance, A. vinelandii displays an expression and transcriptional regulation system of Mo–Fe nitrogenase, encoded by the nif cluster, which includes a major nif cluster and a minor nif cluster. The major nif cluster contains five major gene clusters (nifHDKTY, nifENX, orf5, iscAnifnifUSV–cysE1nif–nifWZM–clpX2, and nifF) encoding the nitrogenase complex. The nifHDKTY gene cluster contains the structural genes of Mo–Fe nitrogenase, which include nifH, nifD, nifK, nifT, and nifY. Moreover, the nifLA operon, located at the minor nif cluster (rnfABCDGEH, nifLAB, fdxN, nifOQ, rhdN, and grx5nif), encodes the NifA protein and NifL protein that regulate and control the transcription of nitrogenase (Figure 1a) [21]. Since the first discovery of the function of the nifLA operon, the research on the NifL–NifA binary regulatory system has achieved significant progress: the function of nitrogen fixation is regulated by the nifLA operon in A. vinelandii, nifA distal, and nifL proximal to the promoter (Figure 1a) [8]. Moreover, the expression of the nifLA gene is rarely affected by environmental factors, and it is continuously expressed or has little change in various growth stages [21][22][21,22]. NifA is the transcriptional activator protein of the nif gene cluster, while NifL inhibits the activity of NifA via interacting with NifA. The NifL–NifA system is affected by the intracellular redox environment and binding state of the ligand (2–OG, ATP/ADP, FAD). The reversible modification of GlnK by uridylation also has a regulatory effect on the transcription of the NifL–NifA system. When there is excess nitrogen, nitrogen metabolism results in low concentrations of 2–OG and high concentrations of glutamine. Glnk, NifA, and NifL can form the ternary complex to suppress the activity of NifA, which results in the blockage of nitrogenase expression. When nitrogen is limited, the floundering nitrogen metabolism results in the accumulation of 2–OG and the excessive consumption of glutamine, which disrupts the formation of the ternary complex to activate NifA. The active NifA can promote the expression of nitrogenase (Figure 2).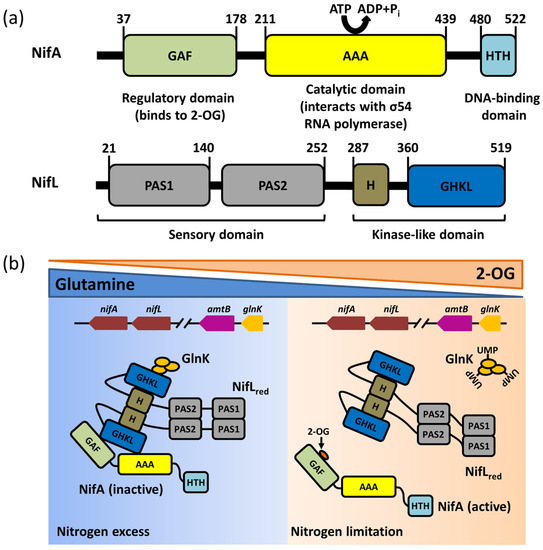
Figure 2. The domain structures and functions of NifA and NifL. (a) Schemes of the domain structures of the NifA and NifL protein; (b) The distinct regulatory modes of NifA–NifL–GlnK complex in the excess or limitation of nitrogen. In nitrogen excess, GHKL domain of NifL protein interacts with NifA protein to inhibit NifA. Glnk can interact with NifL, enhancing the inhibition of NifA activity by NifL. When nitrogen is limited, the uridylylation of Glnk disrupts the interaction between Glnk and NifL. 2–OG can bind to GAF domain of NifA to remove the inhibition of NifA activity by NifL.
3. The NifL–NifA System Responds to the Transcriptional Regulation of Nitrogenase via Environmental Signaling Molecules
The NifL–NifA system of A. vinelandii integrates the intracellular redox and nitrogen and carbon status to regulate the expression of nitrogenase. The interaction between NifL and NifA is regulated in response to the intracellular redox environmental, ligand (2–OG, ATP/ADP, FAD/FADH2) binding status, and the signal–transduction protein GlnK. Under an adverse redox state (excess oxygen) or nitrogen–excess condition, oxidized NifL and NifA form binary complexes to suppress NifA activity. In addition, non–covalently modified GlnK can also interact with NifL to promote the formation of a GlnK–NifL–NifA ternary complex and inhibit NifA activity (Figure 34a). Relatively, in nitrogen–limiting conditions, 2–OG at a high concentration binds to the GAF domain and leads to uridylation of Glnk by GlnD. This can ensure NifA dissociation from the NifL–NifA complex so that free NifA can activate the transcription of the nitrogenase gene (Figure 34b) [23][29].
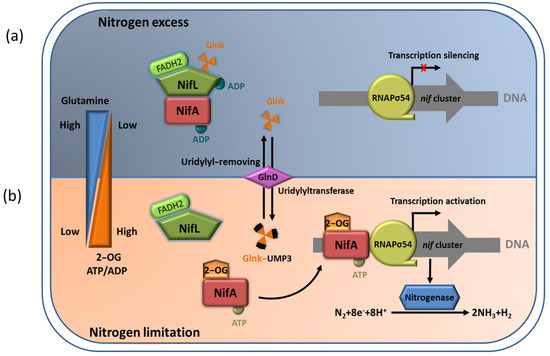
Figure 34. The NifL–NifA system’s responses to environmental and metabolic conditions in A. vinelandii. (a) In nitrogen–excess conditions, the binding of high concentration of glutamine to GlnD results in uridylyl removal from Glnk. NifL forms the binary complexes with NifA to inhibit NifA activation. In addition, the non–covalently modified GlnK can also interact with NifL to promote the formation of the GlnK–NifL–NifA ternary complex and inhibit NifA activity, leading to the transcriptional silencing of nif cluster; (b) In nitrogen–limiting conditions, high concentration of 2–OG can result in the association of NifL and GlnK in the NifA–NifL–Glnk complex. Free NifA can activate the transcription of nitrogenase gene.
3.1. Regulation of NifA Function by 2–OG
As an important intermediate product of the tricarboxylic acid cycle (TCA), 2–OG is considered to be a key signal, which reflects the carbon metabolism status of cells. At the same time, 2–OG also provides the carbon skeleton for nitrogen assimilation, and its concentration indirectly corresponds to the status of intracellular nitrogen [24][41]. In vivo experiments indicated that the physiological concentration of 2–OG increases sharply from about 100 μmol/L to about 1 mmol/L, when the growth condition is changed from a nitrogen–excess state to a nitrogen–limiting state in E. coli [25][42]. In A. vinelandii, 2–OG directly affects the formation of the NifL–NifA complex in a concentration–dependent manner [26][36].
The binding of 2–OG to the GAF domain of NifA can regulate the response of NifA to NifL. The isothermal titration calorimetry (ITC) results showed that both the full–length NifA protein and GAF domain alone could bind 2–OG, and the affinity for either one of them is almost 60 μmol/L. The deletion of the GAF domain loses the ability to bind to 2–OG [27][43]. Limited protease hydrolysis experiments showed that 2–OG bound to the GAF domain increases the sensitivity of the GAF domain to trypsin digestion and inhibits the protection of these digestion sites by NifL [27][43]. This suggests that the binding of 2–OG probably leads to the allosteric reaction of the GAF domain, interrupting the inhibition of NifA by NifL. Consistently, the NifA–F119S mutant in the GAF domain is observed to lose the ability to bind with 2–OG without affecting the ability to bind to NifL, whereas the complex formed by NifA–F119S and NifL is no longer in control of 2–OG [26][36]. These results indicate that the binding of 2–OG to the GAF domain in a concentration–dependent manner induces the conformational changes in the GAF domain, which is followed by dissociation of the NifL–NifA complex, releasing the activity of NifA to activate transcription of the nitrogenase gene and promoting nitrogen fixation.
3.2. Effects of ADP and FAD Molecules on NifL Function
The formation of the NifL–NifA complex is associated with ATP/ADP and FAD/FADH2. The results of affinity chromatography proved that NifA forms a stable complex with NifL at a 1 mmol/L concentration of ADP, whereas the removal of ADP results in complex dissociation [28][44], suggesting a key role in reinforcing the stability of the NifL–NifA complex. ADP stabilizes the NifL–NifA complex by binding to the GHKL domain of NifL, enhancing its inhibitory activity [26][29][30][33,34,36]. The binding of 2–OG to the GAF domain of NifA alters the NifA conformation to antagonize the inhibitory activity of the ADP–bound NifL [26][36]. The catalysis of open promoter complexes by NifA requires hydrolysis of nucleotide triphosphate to supply energy, which is usually provided in the form of ATP or GTP. In vitro transcriptional experiments of open promoter complexes showed that ATP or GTP at a saturating concentration, with 4 mM GTP or 3.5 mM of ATP, can improve the formation of the inhibitory NifL–NifA complex, and the extra–low concentration of ADP (50 μM) can increase inhibition [31][45]. These data support the view that the accumulation of ADP promotes the formation of the NifA–NifL complex during nitrogen fixation, thereby regulating the efficiency of nitrogen fixation.
In the NifL–NifA system, FAD and FADH2 are two signal molecules that manipulate NifA activity via binding to NifL. FAD and FADH2 can reflect the intracellular oxidation/reduction status. The PAS1 domain of NifL can sense the redox status of FAD/FADH2 molecules. In the oxygenated state, the conformational change of NifL produced by the binding of the PAS1 domain to FAD prohibits the activity of NifA upon interacting with it. In addition, NifL resembles the oxidized NifL–NifA complex in the FAD spectral characteristics, regardless of the ADP binding state of NifL. This suggests that the FAD signal rather than ADP determines the inhibition of NifA activity by NifL [31][45]. Relatively, in the reduced state, the binding of the PAS1 domain to FADH2 causes NifL to abolish its ability to interact with NifA [32][46]. Previous studies have reported the crystal structure of the PAS1 domain bound to FAD. The structure showed that the PAS1 domain exists as a dimer in an asymmetric unit, and a novel cavity is formed inside each monomer, which can interact with FAD through salt–bridge, hydrogen–bond, and hydrophobic interactions. This structure supports the idea that hydrogen peroxide released by the oxidizing reaction of FAD can mediate the recognition and transmission of the redox signal [33][34][47,48]. The PAS1 domain complexed with FADH2 is extremely difficult to obtain, since FADH2 is easily oxidized to FAD by oxygen. As a result, the structure of NifL has not been reported in a reduced state.
3.3. The NifL–NifA System Regulated by GlnK
The PII protein, which is widely distributed in bacteria, archaea, and plants, functions as a signal–transduction protein in the regulation of nitrogen fixation [35][49]. Several genes have been verified to encode PII paralogues in proteobacteria, such as glnB, glnK, glnJ, glnY, and glnZ [36][50]. Current evidence indicates that A. vinelandii carries a single gene encoding a protein belonging to the PII family, designated glnK [37][40]. The GlnK protein is a PII–like protein sensing cellular nitrogen signals encoded by the glnK gene, which can participate in the transcriptional regulation of NifL–NifA system through its covalently modified urylation transition in A. vinelandii [38][39][51,52]. The glnK gene is often clustered and co–transcribed with amtB genes encoding a membrane–bound NH4+ channel (AmtB) [36][50]. The expression of the glnK–amtB operon in A. vinelandii is not affected by a fixed nitrogen supply, which is in contrast to other bacteria, such as E. coli and K. pneumoniae [40][53]. The GlnK protein exists in the form of a trimer structurally, which can be reversible and covalently modified by the uridylyltransferase/uridylyl–removing enzyme (UTase/UR) GlnD encoded by glnD, a sensor–regulator that responds to the intracellular glutamine concentration. Each GlnK trimer can be covalently modified by up to three uridine groups. In nitrogen–excess conditions, intracellular nitrogen metabolism is active, and glutamine, as an important nitrogen metabolic intermediate, is accumulated in the intracellular environment. Subsequently, GlnD can exert UR activity to catalyze the deuridine acylation of GlnK via binding to glutamine. The unmodified GlnK is involved in the GlnK–NifL–NifA ternary complex formation that can suppress NifA activity [38][51]. It is possible that the unmodified GlnK does not interact directly with NifA but interacts with the C–terminal GHKL domain of NifL to enhance the inhibition of NifA activity by NifL [41][54]. Under intracellular nitrogen–limiting conditions, the synthesis of glutamine is probably blocked, leading to a decline in glutamine concentration. The lower concentration of glutamine reverses GlnD activity, allowing it to exert UTase activity to catalyze the uridine acylation of GlnK. Consequently, the uridine acylation of GlnK induces conformational changes, abrogating its capability to interact with the NifL–NifA complex [40][42][53,55].
Mutations in GlnD that decrease its activity in the uridine adenylation of Glnk can block the synthesis of nitrogenase via stabilizing the formation of the GlnK–NifL–NifA ternary complex in A. vinelandii [42][55]. In addition, in vitro binding assays proved that unmodified GlnK can promote the formation of the NifA–NifL complex, while uridine–acylated GlnK lost such an ability. These results supported the function of GlnK in regulating the NifL–NifA complex. The previous crystal structure showed that the working mechanism of GlnK is reversibly uridylylated at the conserved Try51 by the GlnD protein in E. coli in the presence of low nitrogen levels. The conserved Try51 is located on the T–loop interacting with the target protein in E. coli, and the uridine acylation at Try51 can enhance the flexibility of the T–loop, which is not conducive to the interaction between Glnk and the target protein [43][56]. This result was further supported by the fact that the GlnK–Y51F mutation failed proper uridine acylation and constant inhibition of NifA activity by NifL [38][51]. Except for reversible modification of uridine acylation, GlnK may suffer from a non–reversible modification in direct response to nitrogen availability. The irreversible modification was identified as specific cleavage of the first three N–terminal amino acids of Glnk in Streptomyces coelicolor, which is speculated to be caused by ammonium shock. However, it has not yet been verified that the specific cleavage of the three N–terminal amino acids can affect the regulation of NifA activity by NifL by modulating GlnK stability [44][57].
The interaction of the PII protein with other proteins is also modulated by the binding of the effectors, including adenylylate energy charge (ADP and ATP) and 2–OG, which regulates signal–transduction proteins, metabolic energy, and permeases involved in nitrogen assimilation [45][46][58,59]. GlnK itself can directly perceive nitrogen limitation in response to 2–OG and ATP/ADP. Moreover, 2–OG, at the appropriate concentration, is a prerequisite for the interaction between unmodified GlnK and NifL in A. vinelandii, consistent with the interaction between PII proteins and other targeted proteins in E. coli [23][47][29,31]. One GlnK trimer in A. vinelandii can bind two to three 2–OG molecules, but 2–OG at high concentration (2 mmol/L) is unable to disrupt the interaction between unmodified GlnK and NifL. This suggests that GlnK is not sensitive to the concentration change of 2–OG within the physiological range in A. vinelandii. Therefore, the regulatory signal of GlnK uridylation is more crucial than 2–OG for GlnK– regulated NifL activity.