Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Gianluca Gaidano and Version 2 by Rita Xu.
Richter syndrome (RS) represents the occurrence of an aggressive lymphoma, most commonly diffuse large B-cell lymphoma (DLBCL), in patients with chronic lymphocytic leukemia (CLL).
- Richter syndrome
- chronic lymphocytic lymphoma
- pathogenesis
- genetic lesions
1. Definition of Richter Syndrome
Richter Syndrome (RS) was reported for the first time by Maurice N. Richter as “reticular cell sarcoma” in 1928 [1]. Currently, according to the World Health Organization (WHO) classification of Tumours of Haematopoietic and Lymphoid Tissues, RS is defined as the occurrence of an aggressive lymphoma in patients with a previous or concomitant diagnosis of chronic lymphocytic leukemia (CLL) or small lymphocytic lymphoma (SLL) [2]. RS is currently divided into two recognized pathological variants: diffuse large B-cell lymphoma (DLBCL) variant, with confluent sheets of large neoplastic post-germinal center B lymphocytes, and Hodgkin lymphoma (HL) variant [2][3][2,3].
The neoplastic cells of DLBCL-type RS express CD20, and less commonly CD5 and CD23 [2]. PD-1 expression is documented in up to 80% of DLBCL-type RS, whereas in de novo DLBCL, this marker is poorly expressed. Another difference between DLBCL-type RS and de novo DLBCL is the low rate of BCL2 genetic lesions, compared with the prevalence of BCL2 translocations and somatic mutations that are commonly found in de novo DLBCL [4][5][6][4,5,6]. The analysis of immunoglobulin genes has shown that ~80% of the cases of DLBCL-type RS are clonally related to the CLL phase, thus documenting that this histologic shift is a true transformation event from the previous indolent phase. However, a minority (~20%) of DLBCL-type RS cases are characterized by a rearrangement of immunoglobulin genes that is distinct from that of the CLL phase, documenting a clonally unrelated origin of RS [3].
The HL variant is similar to its de novo counterpart, with the presence of Hodgkin and Reed–Sternberg cells in a typical background of reactive T cells, epithelioid histiocytes, eosinophils and plasma cells or, eventually, interspersed in a background of CLL cells [7][8][9][7,8,9]. Hodgkin and Reed–Sternberg cells are characterized by a CD30+/CD15+/CD20− immunophenotype and are often found to be EBV positive [3][9][3,9].
2. Epidemiology
CLL is the most frequent leukemia in adults, with an incidence of 4.7/100,000 per year in the US [10]. In the chemo-immunotherapy (CIT) era, data mainly based on retrospective studies showed an incidence of RS transformation ranging from 1 to 7% [11]. Consistently, the CALGB 9011 clinical trial demonstrated a RS transformation rate of ~7% in treatment-naïve CLL patients after at least 15 years after treatment with fludarabine or chlorambucil [12]. Remarkably, in the CLL8 trial evaluating the effect of fludarabine and cyclophosphamide with or without the anti-CD20 monoclonal antibody (mAb) rituximab, the use of rituximab proved to be a protective factor against RS transformation, leading to lower rates of progression to RS in patients receiving rituximab [13]. A retrospective study suggested a potential role played by prolymphocytes in RS development, underlining that deaths due to RS were significantly more common in CLL patients who had ≥10% circulating prolymphocytes [14].
More recently, a pooled analysis of the German CLL Study Group (GCLLSG) considering frontline treatment trials with both CIT and pathway inhibitors, including Bruton tyrosine kinase (BTK) and BCL2 inhibitors, has documented a 3% prevalence of RS transformation among 2975 CLL patients monitored after their enrolment in clinical trials, recruited from 1999 to 2016, with a median observation time of 53 months [15]. Data from the Surveillance, Epidemiology and End Results (SEER) database of CLL patients diagnosed between 2000 and 2016 have documented that the incidence of RS transformation was 0.7% [16].
The issue of RS epidemiology in the era of novel agents has been partially answered by results collected from the first clinical trials with pathway inhibitors: in first-line treatment, novel agents showed RS transformation rates comparable to those of the CIT era, suggesting that pathway inhibitors are neither harmful nor fully protective [17][18][17,18]. Among relapsed/refractory (R/R) CLL patients treated with novel agents, the RS incidence was higher than the overall incidence of RS in the CIT era, probably due to the biological behavior and genetic profile of R/R CLL [19][20][21][22][19,20,21,22]. Because pathway inhibitors have been approved for the treatment of CLL relatively recently (the first BTK inhibitor, ibrutinib, was approved in 2014), further investigation is needed to assess the RS transformation rate with these novel agents in a real-life setting. Currently available data on RS frequency in cases treated with pathway inhibitors are summarized in Table 1.
Table 1. RS frequency in patients treated with therapeutic regimens based on pathway inhibitors.
| Number of CLL Patients | Study Population | Treatment | RS Prevalence (%) | Reference |
|---|---|---|---|---|
| 391 | Relapsed | Ibrutinib, ofatumumab | 1 | Byrd, 2014 [23] |
| 29 | Progressive untreated | Ibrutinib | 3 | O’Brien, 2014 [24] |
| 194 | R/R | Venetoclax-rituximab | 3 | Seymour, 2018 [19] |
| 127 | R/R | Ibrutinib | 5 | Jain, 2015 [25] |
| 84 | 17p deleted or ≥65 years | Ibrutinib | 6 | Ahn, 2017 [21] |
| 358 | Treatment-naïve | Acalabrutinib, Obinutuzumab | 2 | Sharman, 2020 [18] |
| 51 | 17p deleted | Ibrutinib | 6 | Farooqui, 2015 [26] |
| 178 | BCRi treated | Ibrutinib, idelalisib | 7 | Mato, 2016 [27] |
| 113 | Treatment-naïve | Ibrutinib-obinutuzumab | 0 | Moreno, 2019 [17] |
| 85 | R/R | Ibrutinib | 8 | Byrd, 2013 [28] |
| 116 | R/R | Venetoclax | 16 | Roberts, 2016 [29] |
| 67 | R/R, 17p deleted | Venetoclax | 25 | Anderson, 2017 [30] |
| 2975 | R/R | B, F, C, Clb, rituximab, obinutuzumab, ibrutinib, venetoclax | 3 | Al-Sawaf, 2021 [15] |
| 195 | R/R | Ibrutinib | 10 | Munir, 2019 [20] |
3. Molecular Pathways in RS
Several molecular alterations associated with DLBCL-type RS have been described, whereas the development of HL-type RS has been studied less extensively and is thought to be similar to that of de novo HL and possibly linked to EBV-mediated immunosuppression, thus favoring CLL progression to HL [7][31][32][7,31,32].
Genetic lesions of RS. The pathogenesis of DLBCL-type RS is linked to the dysregulation of intracellular pathways involved in DNA damage response, tumor suppression, apoptosis, modulation of the cell cycle and proliferation. The main genetic lesions associated with DLBCL-type RS are represented by somatic mutations or disruptions of the TP53, CDKN2A, NOTCH1 and c-MYC genes (Figure 1) [2][5][6][7][33][34][2,5,6,7,33,34].
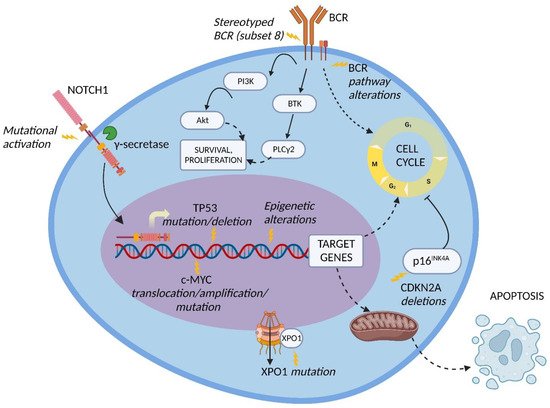
Figure 1. Molecular pathway alterations in DLBCL-type RS. The pathogenesis of DLBCL-type RS is due to the dysregulation of multiple molecular pathways due to genetic lesions of proto-oncogenes and tumor suppressor genes, stereotyped B-cell receptor (BCR) configuration, and BCR signaling alterations; these lead to the enhanced cell survival and proliferation typical of DLBCL-type RS cells. Inhibition of apoptosis may also be involved. BTK, Bruton tyrosine kinase; PI3K, phosphatidylinositol-3 kinase; PLCγ2, phospholipase C gamma 2; XPO1, exportin 1. Image created with Biorender.com (accessed on 4 August 2022).
TP53 encodes for one of the main regulators of the DNA-damage-response pathway, and its disruption, generally acquired at the time of transformation, leads to the chemorefractoriness characteristic of RS and, therefore, favors the positive selection and expansion of mutated tumor cells [34][35][34,35]. Disruption of TP53 by mutation and/or deletion has been documented in a large fraction of DLBCL-type RS, including 60 to 80% of clonally related RS cases, which represent the overwhelming majority of RS events, and in 20% of clonally unrelated RS cases [6]. The high recurrence of TP53 disruption explains, at least in part, the frequency of chemorefractoriness in this condition.
CDKN2A deletion occurs in ~30% of DLBCL-type RS and is commonly acquired at the time of transformation [5]. The CDKN2A gene is responsible for the negative regulation of the G1 to S transition of the cell cycle and for the activation of the p53 transcriptional program through its transcripts p16INK4A and p14ARF, respectively, leading to tumor suppression [5][34][36][5,34,36]. Importantly, both positive and negative cell cycle regulators are induced by B-cell receptor (BCR) signaling in murine models. On these grounds, the concomitant loss of the negative cell cycle regulators TP53 and CDKN2A/B shifts BCR-dependent signaling toward the promotion of positive cell cycle regulators, leading to an aggressive proliferation compatible with RS transformation [37].
NOTCH1 encodes for a surface receptor that, after being triggered by a ligand belonging to the SERRATE/JAGGED or DELTA families, is cleaved by γ-secretase, migrates into the nucleus and activates the transcription of several genes involved in cell proliferation and survival [38]. At the time of diagnosis, the frequency of NOTCH1 mutational activation in RS is significantly higher compared to the frequency in CLL (31% vs. 8.3%, respectively) [39]. These data, together with the noticeably higher reported risk of developing DLBCL-type RS in NOTCH1 mutated CLL (45% at 15 years) vs. NOTCH1 wild type CLL (4.6% at 15 years), indicate that mutations of NOTCH1 are a significant risk factor for developing RS transformation [40].
The c-MYC proto-oncogene and its transcriptional product take part in many crucial cellular pathways, and a dysregulation of this network results in altered cell survival, proliferation, metabolism, self-renewal, and genomic instability [41]. The main genetic lesions deregulating c-MYC expression in DLBCL-type RS are represented by chromosomal translocations between the c-MYC locus and the IGHV regulatory regions, and gene amplifications as well as gain-of-function mutations of the c-MYC promoter. An alternative mechanism of c-MYC deregulation is represented by loss-of-function mutations of the MGA gene, which encodes for a protein inhibiting c-MYC heterodimerization with its partner MAX. Overall, by combining genetic lesions of c-MYC and MGA, ~40% of DLBCL-type RS cases harbor c-MYC deregulation [42].
The NF-κB pathway is also involved in RS pathogenesis [43]. TRAF3, a gene implicated in the negative regulation of signaling through the NF-κB and mitogen-activated protein kinase (MAPK) pathways, is disrupted by heterozygous deletions and frameshift mutations in a fraction of RS cases [43]. Inactivation of TRAF3 leads to NF-κB activation, promoting B cell survival and, in particular, enhancing the expression of c-MYC and PIM-2 [44]. PIM-2 maintains high levels of NF-κB, which are required for its antiapoptotic function. The pathogenetic role of PIM-2 in B-cell neoplasia is documented by its overexpression, translocation, or amplification in a fraction of B-cell lymphomas [43][44][45][46][43,44,45,46].
Other molecular alterations detected in DLBCL-type RS include (i) overexpression and amplification of PTPN11, a positive regulator of the MAPK-RAS-ERK signaling pathway; (ii) deletion of the SETD2 histone methyltransferase, which plays a major role in chromatin epigenetic remodeling; and (iii) disrupting mutations of the tumor suppressor gene PTPRD, which encodes for a receptor-type protein, tyrosine phosphatase, regulating cell growth and found to be inactivated also in other types of B-cell neoplasia and solid cancers [43][47][48][49][50][43,47,48,49,50].
Modification of immune regulators in RS. Recent studies have underlined the importance of immune checkpoints and of the tumor microenvironment in lymphatic tissues of DLBCL-type RS [4][51][4,51]. The main immune checkpoints involved in RS are PD-1, LAG3 and TIGIT.
PD-1 is a T cell surface molecule which stimulates effector T-cell apoptosis and Treg survival through its binding with the PD-L1 ligand, which is expressed mainly on the surface of antigen-presenting cells (APC), such as macrophages, B cells and dendritic cells (DC) [52]. Augmented levels of PD-1 in RS cells and enhanced expression of PD-L1 in histiocytes and dendritic cells of the RS microenvironment have been reported [4][51][4,51]. Altered expression of the PD-1/PD-L1 axis leads to RS tumor-cell resistance to the cytotoxicity exerted by T cells [52].
LAG3 is a T cell surface protein with a structure similar to the T helper antigen CD4, and its main ligand is the major histocompatibility complex (MHC) class II, typically expressed by APC. Additional ligands have been identified, namely Galectin-3 (Gal-3), which is expressed by several cell types [53]. The interaction between LAG3 and its ligands promotes tumor escape from apoptosis through the recruitment of tumor-specific CD4+ T cells (through the interaction with MHC class II) and the inhibition of CD8+ T cells’ cytotoxic function by Gal-3 binding [53]. Higher LAG3 levels have been observed in RS neoplastic and tumor-infiltrating lymphocytes, suggesting its potential role in promoting tumor immune escape and neoplastic cell survival [54].
TIGIT, expressed on normal T and NK cells and overexpressed in RS, is capable of immune suppression as a consequence of its binding with the CD155 ligand, exposed on the cell membrane of various cell types, such as dendritic cells, T cells, B cells, and macrophages [54][55][54,55]. The mechanism of action of TIGIT is supposed to be linked to the transduction of immune-suppressive stimuli on T and NK cells and to the promotion of tolerogenic DC that downregulate T cell responses [55]. The finding that immune checkpoints are overexpressed in RS cells and tumor-infiltrating lymphocytes suggests the potential role of these molecules in the promotion of a permissive immune microenvironment, resulting in immune suppression and tumor escape.
The BCR pathway in RS. Several BCR pathway alterations related to RS transformation have been documented. The BCR is a transmembrane complex expressed in B cells, composed by a surface immunoglobulin linked to a signal transduction subunit and responsible for antigen recognition and B cell activation (Figure 1) [56].
The variable part of the BCR IGHV subunit is characterized by a molecular pattern typical of mature B cells, the VDJ rearrangement, which causes a considerable diversity across the BCR expressed by different B cell clones. Approximately thirty percent of CLL patients carry stereotyped BCR, which are characterized by almost identical VDJ rearrangement across patients and are groupable in well-codified subsets identified by progressive numbers [57][58][57,58].
CLL patients carrying BCR subset 8 (characterized by IGHV4-39/IGHD6-13/IGHJ5 rearrangement) display a significantly higher risk of developing DLBCL-type RS, especially in combination with NOTCH1 mutations. [59][60][59,60]. From a mechanistic perspective, CLL cells harboring BCR subset 8 tend to overreact to multiple autoantigens and immune stimuli derived from the microenvironment (Figure 1). The propensity of these cells to undergo RS transformation can be explained by this promiscuous antigen reactivity [61].
In the BCR signal transduction, a key role is played by Bruton tyrosine kinase (BTK) and phosphatidylinositol-3 kinase (PI3K) [62]. BTK is phosphorylated subsequently to BCR stimulation and leads to the activation of phospholipase C gamma 2 (PLCγ2), causing calcium mobilization and activation of cell survival, proliferation and differentiation pathways, including MAPK and NF-κB signaling (Figure 1) [63]. PI3K, which is responsible for the activation of the serine/threonine kinase Akt and for the delta isoform of protein kinase C (PKC), was observed to be constitutively active in CLL patients, resulting in an enhanced anti-apoptotic effect [64].
Akt signaling in RS. Akt takes part in cell-survival signaling through mTOR and is constitutively active in high-risk CLL (i.e., TP53 or NOTCH1 mutated CLL) and in >50% of cases of RS [64][65][64,65]. In an Eµ-TCL1 murine model of CLL with constitutively active Akt alleles in B cells, the excessive Akt activation led to an aggressive DLBCL-type lymphoma with histological and biological features coherent with human RS [65]. Additionally, this murine model enlightened the correlation between hyperactivation of Akt and NOTCH1 signaling, since mice with constitutively active Akt alleles presented an expansion of CD4+ T cells expressing the NOTCH1 ligand DLL1 in the tumor microenvironment, implying a higher engagement of NOTCH1 by its ligands in neoplastic cells [65].