Low back pain is the leading cause of disability worldwide, with up to 80% of the population experiencing this at some point in their life. Chronic back pain is complex and there is currently no cure for it, nor does alleviating potential causes guarantee that pain will go away. Therefore, rather than attempting to “cure” chronic pain, many clinicians, caregivers and researchers aim to help educate patients about their pain and try to help them live a better quality of life despite their condition. This may include using strategies such as pain neuroscience education (PNE) and cognitive behavior therapy. PNE, for example, is considered an intervention aimed at reconceptualizing an individual’s understanding of their pain as less threatening. A systematic review and meta-analysis demonstrated that PNE can have a significant effect in reducing pain catastrophizing as well as kinesiophobia. This is highly beneficial in pain management, as reduced catastrophic thinking can help orient a person away from their pain and towards living their life, and reduced fear helps patients to be more open to active interventions like physical therapy and exercise. Pain education is often paired with physical therapy, or used pre-emptively for things like post-operative pain.
- chronic back pain
- spine
- cLBP
1. The Pathophysiology of Back Pain
1.1. Summary of Pathophysiology
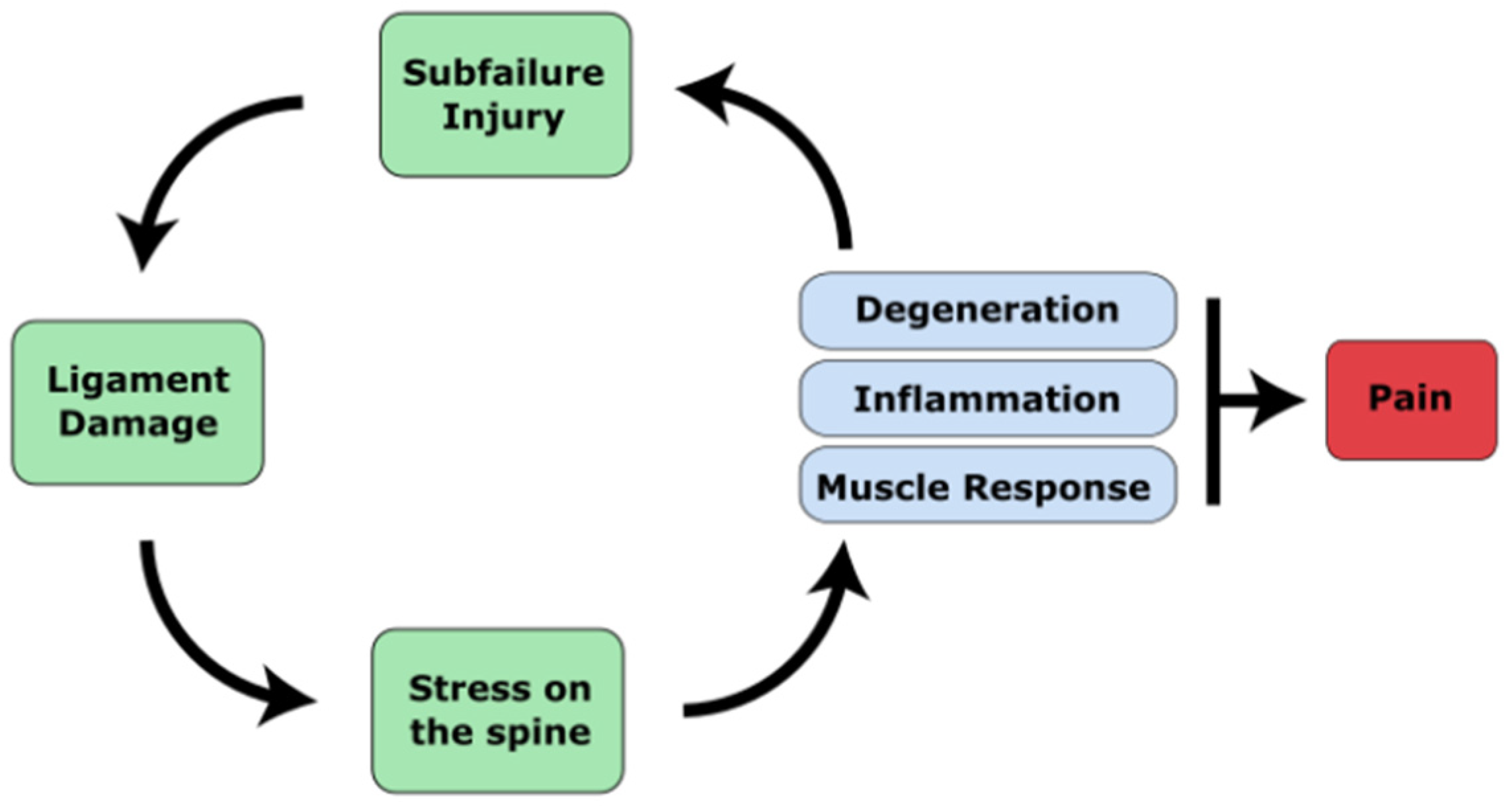
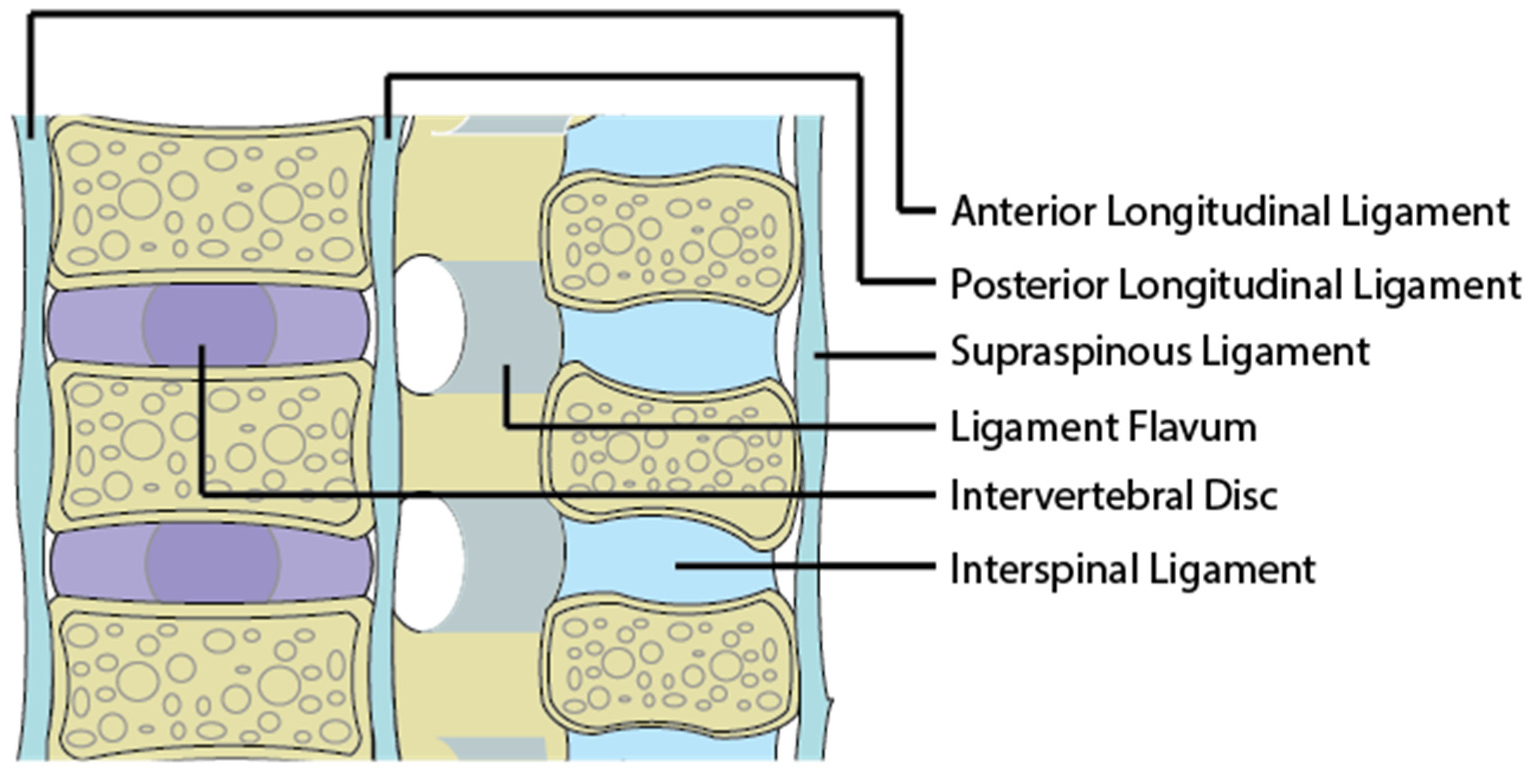
1.2. The Ligamentous Concept
1.3. Myofascia and Spinal Muscles
1.4. Spinal Muscles and Mechanoreceptors
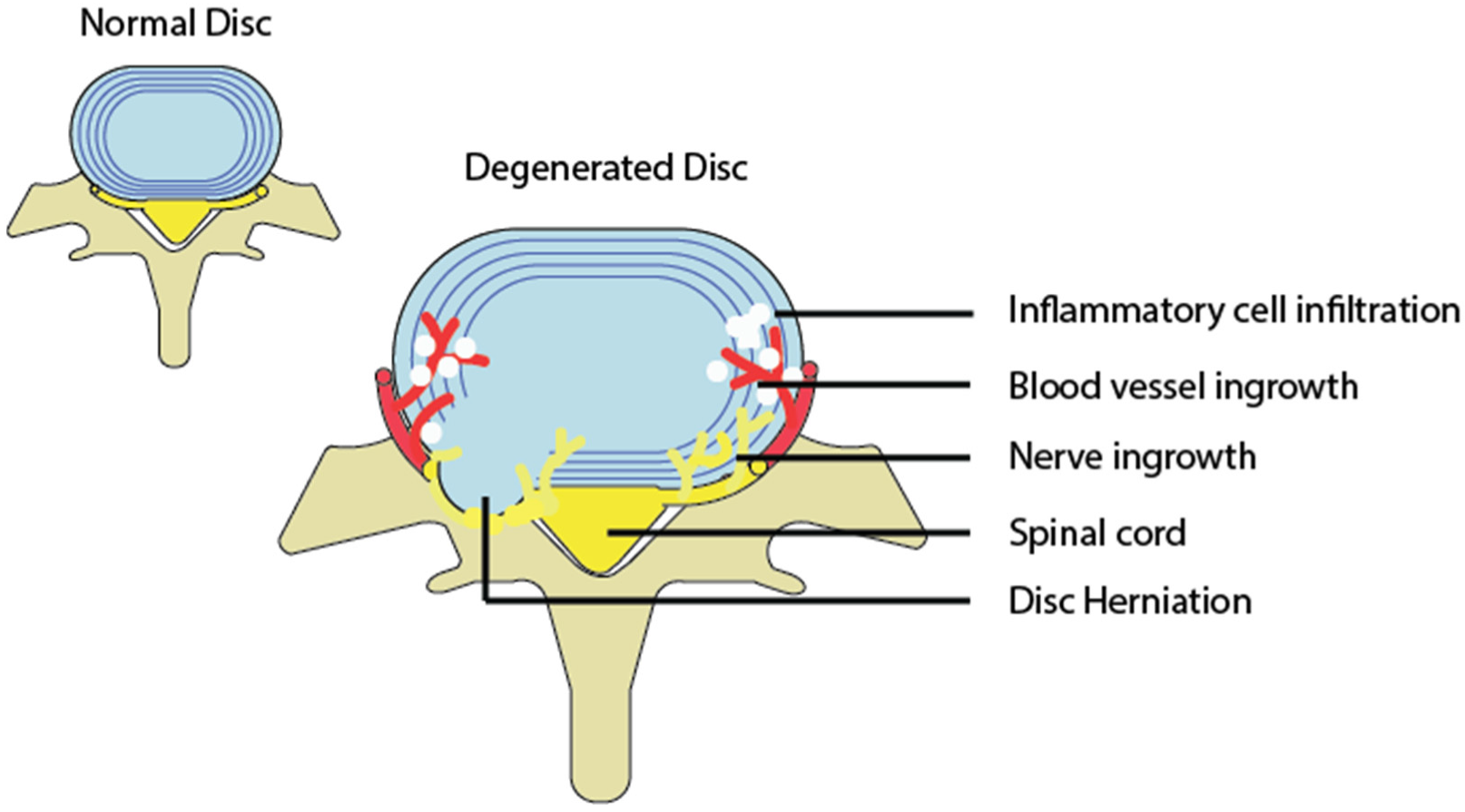
1.5. Degenerative Discs and the Inflammatory Response
1.6. Spinal Osteoarthritis and Degenerative Joint Disease
2. The Transition from Acute to Chronic Pain
References
- Waddell, G. 1987 volvo award in clinical sciences: A new clinical model for the treatment of low-back pain. Spine 1987, 12, 632–644.
- Mannion, A.F.; Dolan, P.; Adams, M.A. Psychological questionnaires: Do “abnormal” scores precede or follow first-time low back pain? Spine 1996, 21, 2603–2611.
- Adams, M.A. Biomechanics of back pain. Acupunct. Med. 2004, 22, 178–188.
- Panjabi, M.M. A hypothesis of chronic back pain: Ligament subfailure injuries lead to muscle control dysfunction. Eur. Spine J. 2006, 15, 668–676.
- Allegri, M.; Montella, S.; Salici, F.; Valente, A.; Marchesini, M.; Compagnone, C.; Baciarello, M.; Manferdini, M.E.; Fanelli, G. Mechanisms of low back pain: A guide for diagnosis and therapy. F1000Research 2016, 5, 1530.
- Urits, I.; Burshtein, A.; Sharma, M.; Testa, L.; Gold, P.A.; Orhurhu, V.; Viswanath, O.; Jones, M.R.; Sidransky, M.A.; Spektor, B.; et al. Low Back Pain, a Comprehensive Review: Pathophysiology, Diagnosis, and Treatment. Curr. Pain Headache Rep. 2019, 23, 23.
- Senington, B.; Lee, R.Y.; Williams, J.M. Biomechanical risk factors of lower back pain in cricket fast bowlers using inertial measurement units: A prospective and retrospective investigation. BMJ Open Sport—Exerc. Med. 2020, 6, 818.
- Li, W.; Gong, Y.; Liu, J.; Guo, Y.; Tang, H.; Qin, S.; Zhao, Y.; Wang, S.; Xu, Z.; Chen, B. Peripheral and Central Pathological Mechanisms of Chronic Low Back Pain: A Narrative Review. J. Pain Res. 2021, 14, 1483.
- Rubinstein, S.M.; van Tulder, M. A best-evidence review of diagnostic procedures for neck and low-back pain. Best Pract. Res. Clin. Rheumatol. 2008, 22, 471–482.
- Donnelly, J.M. Travell, Simons & Simons’ myofascial pain and dysfunction : The trigger point manual. Wolters Kluwer Health 2019, xxvii, 935.
- Vleeming, A.; Pool-Goudzwaard, A.L.; Stoeckart, R.; van Wingerden, J.P.; Snijders, C.J. The Posterior Layer of the Thoracolumbar.pdf. Spine J. 1995, 20, 753–758.
- Barker, P.J.; Briggs, C.A. Attachments of the posterior layer of lumbar fascia. Spine 1999, 24, 1757–1764.
- Schleip, R.; Vleeming, A.; Lehmann-Horn, F.; Klingler, W. Letter to the Editor concerning “A hypothesis of chronic back pain: Ligament subfailure injuries lead to muscle control dysfunction” (M. Panjabi) . Eur. Spine J. 2007, 16, 1733–1735.
- Hodges, P.W.; Danneels, L. Changes in Structure and Function of the Back Muscles in Low Back Pain: Different Time Points, Observations, and Mechanisms. J. Orthop. Sports Phys. Ther. 2019, 49, 464–476.
- Claus, A.P.; Hides, J.A.; Moseley, G.L.; Hodges, P.W. Different ways to balance the spine in sitting: Muscle activity in specific postures differs between individuals with and without a history of back pain in sitting. Clin. Biomech. 2018, 52, 25–32.
- Karayannis, N.V.; Smeets, R.J.E.M.; van den Hoorn, W.; Hodges, P.W. Fear of Movement Is Related to Trunk Stiffness in Low Back Pain. PLoS ONE 2013, 8, e67779.
- Mannion, A.F.; Käser, L.; Weber, E.; Rhyner, A.; Dvorak, J.; Müntener, M. Influence of age and duration of symptoms on fibre type distribution and size of the back muscles in chronic low back pain patients. Eur. Spine J. 2000, 9, 273–281.
- Özcan-Ekşi, E.E.; Ekşi, M.Ş.; Turgut, V.U.; Canbolat, Ç.; Pamir, M.N. Reciprocal relationship between multifidus and psoas at L4-L5 level in women with low back pain. Br. J. Neurosurg. 2021, 35, 220–228.
- Özcan-Ekşi, E.E.; Ekşi, M.Ş.; Akçal, M.A. Severe Lumbar Intervertebral Disc Degeneration Is Associated with Modic Changes and Fatty Infiltration in the Paraspinal Muscles at all Lumbar Levels, Except for L1-L2: A Cross-Sectional Analysis of 50 Symptomatic Women and 50 Age-Matched Symptomatic Men. World Neurosurg. 2019, 122, e1069–e1077.
- Kojima, Y.; Maeda, T.; Arai, R.; Shichikawa, K. Nerve supply to the posterior longitudinal ligament and the intervertebral disc of the rat vertebral column as studied by acetylcholinesterase histochemistry. II. Regional differences in the distribution of the nerve fibres and their origins. J. Anat. 1990, 169, 247–255.
- Sekine, M.; Yamashita, T.; Takebayashi, T.; Sakamoto, N.; Minaki, Y.; Ishii, S. Mechanosensitive Afferent Units in the Lumbar Posterior Longitudinal Ligament. Spine 2001, 26, 1516–1521.
- Cavanaugh, J.M.; Ozaktay, A.C.; Yamashita, T.; Avramov, A.; Getchell, T.V.; King, A.I. Mechanisms of low back pain: A neurophysiologic and neuroanatomic study. Clin. Orthop. Relat. Res. 1997, 335, 166–180.
- McLain, R.F. Mechanoreceptor Endings in Human Cervical Facet Joints. Spine 1994, 19, 495–501.
- Yamashita, T.; Cavanaugh, J.M.; el-Bohy, A.A.; Getchell, T.V.; King, A.I. Mechanosensitive afferent units in the lumbar facet joint. J. Bone Jt. Surg. Am. 1990, 72, 865–870.
- Solomonow, M.; Zhou, B.; Baratta, R.V.; Zhu, M.; Lu, Y. Neuromuscular disorders associated with static lumbar flexion: A feline model. J. Electromyogr. Kinesiol. 2002, 12, 81–90.
- Solomonow, M.; Zhou, B.-H.; Harris, M.; Lu, Y.; Baratta, R.V. The Ligamento-Muscular Stabilizing System of the Spine. Spine 1998, 23, 2552–2562.
- Indahl, A.; Kaigle, A.M.; Reikerås, O.; Holm, S.H. Interaction between the Porcine Lumbar Intervertebral Disc, Zygapophysial Joints, and Paraspinal Muscles. Spine 1997, 22, 2834–2840.
- Solomonow, M.; Zhou, B.-H.; Baratta, R.V.; Burger, E. Biomechanics and electromyography of a cumulative lumbar disorder: Response to static flexion. Clin. Biomech. 2003, 18, 890–898.
- Williams, M.; Solomonow, M.; Zhou, B.H.; Baratta, R.V.; Harris, M. Multifidus Spasms Elicited by Prolonged Lumbar Flexion. Spine 2000, 25, 2916–2924.
- Roberts, S.; Eisenstein, S.M.; Menage, J.; Evans, E.H.; Ashton, I.K. Mechanoreceptors in intervertebral discs. Morphology, distribution, and neuropeptides. Spine 1995, 20, 2645–2651.
- McLain, R.F.; Pickar, J.G. Mechanoreceptor Endings in Human Thoracic and Lumbar Facet Joints. Spine 1998, 23, 168–173.
- Malmberg, A.B.; Chaplan, S.R. Mechanisms and Mediators of Neuropathic Pain; Birkhäuser: Basel, Switzerland, 2002; ISBN 978-3-0348-9448-7.
- Michaelis, M.; Blenk, K.H.; Janig, W.; Vogel, C. Development of spontaneous activity and mechanosensitivity in axotomized afferent nerve fibers during the first hours after nerve transection in rats. J. Neurophysiol. 1995, 74, 1020–1027.
- Roza, C.; Laird, J.M.A.; Souslova, V.; Wood, J.N.; Cervero, F. The tetrodotoxin-resistant Na+ channel Nav1.8 is essential for the expression of spontaneous activity in damaged sensory axons of mice. J. Physiol. 2003, 550, 921–926.
- Cornefjord, M.; Olmarker, K.; Otani, K.; Rydevik, B. Nucleus pulposus-induced nerve root injury: Effects of diclofenac and ketoprofen. Eur. Spine J. 2002, 11, 57–61.
- Solomonow, M.; Zhou, B.-H.; Baratta, R.V.; Lu, Y.; Harris, M. 1999 Volvo Award Winner in Biomechanical Studies. Spine 1999, 24, 2426.
- Butler, D.; Trafimow, J.H.; Andersson, G.B.J.; MCNEILL, T.W.; Huckman, M.S. Discs Degenerate Before Facets. Spine 1990, 15, 111–113.
- Sampara, P.; Banala, R.R.; Vemuri, S.K.; Av, G.R.; Gpv, S. Understanding the molecular biology of intervertebral disc degeneration and potential gene therapy strategies for regeneration: A review. Gene Ther. 2018, 25, 67–82.
- De Geer, C.M. Cytokine Involvement in Biological Inflammation Related to Degenerative Disorders of the Intervertebral Disk: A Narrative Review. J. Chiropr. Med. 2018, 17, 54–62.
- Adams, M.A.; McNally, D.S.; Dolan, P. “Stress” distributions inside intervertebral discs. The effects of age and degeneration. J. Bone Jt. Surg. Br. 1996, 78, 965–972.
- Luoma, K.; Riihimäki, H.; Luukkonen, R.; Raininko, R.; Viikari-Juntura, E.; Lamminen, A. Low back pain in relation to lumbar disc degeneration. Spine 2000, 25, 487–492.
- Fardon, D.F.; Milette, P.C. Combined Task Forces of the North American Spine Society, American Society of Spine Radiology, and American Society of Neuroradiology Nomenclature and Classification of Lumbar Disc Pathology. Spine 2001, 26, E93–E113.
- Solovieva, S.; Lohiniva, J.; Leino-Arjas, P.; Raininko, R.; Luoma, K.; Ala-Kokko, L.; Riihimäki, H. COL9A3 Gene Polymorphism and Obesity in Intervertebral Disc Degeneration of the Lumbar Spine: Evidence of Gene-Environment Interaction. Spine 2002, 27, 2691–2696.
- Videman, T.; Battié, M.C.; Gill, K.; Manninen, H.; Gibbons, L.E.; Fisher, L.D. Magnetic Resonance Imaging Findings and Their Relationships in the Thoracic and Lumbar Spine. Spine 1995, 20, 928–935.
- Lowrey, J.J. Dislocated lumbar vertebral epiphysis in adolescent children. J. Neurosurg. 1973, 38, 232–234.
- Freemont, A.J.; Peacock, T.E.; Goupille, P.; Hoyland, J.A.; O’Brien, J.; Jayson, M.I. Nerve ingrowth into diseased intervertebral disc in chronic back pain. Lancet 1997, 350, 178–181.
- Freemont, A.J.; Watkins, A.; Le Maitre, C.; Baird, P.; Jeziorska, M.; Knight, M.T.N.; Ross, E.R.S.; O’Brien, J.P.; Hoyland, J.A. Nerve growth factor expression and innervation of the painful intervertebral disc. J. Pathol. 2002, 197, 286–292.
- Wheaton, M.T.; Jensen, N. The Ligament Injury Connection to Osteoarthritis? J. Prolotherapy 2010, 1, 294–304.
- El-Bohy, A.A.; Goldberg, S.J.K.A. Measurement of facet capsular stretch. In Proceedings American Society of Mechanical Engineers Applied Mechanics, Bioengineering, and Fluids Engineering Conference, Cincinnati, OH, USA, 14–17 June 1987.
- Yang, K.H.; King, A.I. Mechanism of facet load transmission as a hypothesis for low-back pain. Spine 1984, 9, 557–565.
- Sims-Williams, H.; Jayson, M.I.V.; Baddeley, H. Small spinal fractures in back pain patients. Ann. Rheum. Dis. 1978, 37, 262–265.
- Lawrence, J.S.; Bremner, J.M.; Bier, F. Osteo-arthrosis. Prevalence in the population and relationship between symptoms and x-ray changes. Ann. Rheum. Dis. 1966, 25, 1–24.
- Adams, M.A.; Hutton, W.C. The mechanical function of the lumbar apophyseal joints. Spine 1983, 8, 327–330.
- Cavanaugh, J.M.; Ozaktay, A.C.; Yamashita, H.T.; King, A.I. Lumbar facet pain: Biomechanics, neuroanatomy and neurophysiology. J. Biomech. 1996, 29, 1117–1129.
- Hirsch, C.; Ingelmark, B.E.; Miller, M. The anatomical basis for low back pain. Studies on the presence of sensory nerve endings in ligamentous, capsular and intervertebral disc structures in the human lumbar spine. Acta Orthop. Scand. 1963, 33, 1–17.
- Marks, R.C.; Houston, T.; Thulbourne, T. Facet joint injection and facet nerve block: A randomised comparison in 86 patients with chronic low back pain. Pain 1992, 49, 325–328.
- McCall, I.W.; Park, W.M.; O’Brien, J.P. Induced pain referral from posterior lumbar elements in normal subjects. Spine 1979, 4, 441–446.
- Popert, A.J. Joint Ligament Relaxation treated by Fibro-Osseous Proliferation. Ann. Rheum. Dis. 1956, 15, 373.
- Goldthwait, J.E. The Lumbo-Sacral Articulation; An Explanation of Many Cases of “Lumbago”, “Sciatica” and Paraplegia. Bost. Med. Surg. J. 1911, 164, 365–372.
- Igarashi, A.; Kikuchi, S.; Konno, S.; Olmarker, K. Inflammatory cytokines released from the facet joint tissue in degenerative lumbar spinal disorders. Spine 2004, 29, 2091–2095.
- Nijs, J.; Lahousse, A.; Kapreli, E.; Bilika, P.; Saraçoğlu, İ.; Malfliet, A.; Coppieters, I.; De Baets, L.; Leysen, L.; Roose, E.; et al. Nociplastic Pain Criteria or Recognition of Central Sensitization? Pain Phenotyping in the Past, Present and Future. J. Clin. Med. 2021, 10, 10.
- Coluzzi, F.; Fornasari, D.; Pergolizzi, J.; Romualdi, P. From acute to chronic pain: Tapentadol in the progressive stages of this disease entity. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 1672–1683.
- D’Mello, R.; Dickenson, A.H. Spinal cord mechanisms of pain. Br. J. Anaesth. 2008, 101, 8–16.
- Chapman, C.R.; Tuckett, R.P.; Song, C.W. Pain and stress in a systems perspective: Reciprocal neural, endocrine, and immune interactions. J. Pain 2008, 9, 122–145.
- Feizerfan, A.; Sheh, G. Transition from acute to chronic pain. Contin. Educ. Anaesth. Crit. Care Pain 2015, 15, 98–102.
- Graven-Nielsen, T.; Arendt-Nielsen, L. Peripheral and central sensitization in musculoskeletal pain disorders: An experimental approach. Curr. Rheumatol. Rep. 2002, 4, 313–321.
- Macintyre, E.P.; Scott, D.A.; Schug, S.A. Acute Pain Management: Scientific Evidence, 3rd ed.; Australian and New Zealand College of Anaesthetists: West End, Australia, 2010.
- Mifflin, K.A.; Kerr, B.J.; Mifflin, K.A.; Kerr, Á.B.J. The transition from acute to chronic pain: Understanding how different biological systems interact. La transition d’une douleur aiguë vers une douleur chronique-comprendre l’interaction entre différents systèmes biologiques. Can. J. Anesth./J. Can. D'anesthésie 2014, 61, 112–122.
- Voscopoulos, C.; Lema, M. When does acute pain become chronic? BJA Br. J. Anaesth. 2010, 105, i69–i85.
- Watkins, L.R.; Hutchinson, M.R.; Rice, K.C.; Maier, S.F. The “toll” of opioid-induced glial activation: Improving the clinical efficacy of opioids by targeting glia. Trends Pharmacol. Sci. 2009, 30, 581–591.
- Walton, K.D.; Llinás, R.R. Central pain as a thalamocortical dysrhythmia: A thalamic efference disconnection? In Translational Pain Research: From Mouse to Man; CRC Press: Boca Raton, FL, USA, 2009; pp. 301–314. ISBN 9781439812105.
- Ahmadi, S.; Lippross, S.; Neuhuber, W.L.; Zeilhofer, H.U. PGE2 selectively blocks inhibitory glycinergic neurotransmission onto rat superficial dorsal horn neurons. Nat. Neurosci. 2002, 5, 34–40.
- Geha, P.Y.; Baliki, M.N.; Chialvo, D.R.; Harden, R.N.; Paice, J.A.; Apkarian, A. V Brain activity for spontaneous pain of postherpetic neuralgia and its modulation by lidocaine patch therapy. Pain 2007, 128, 88–100.
- Malinen, S.; Vartiainen, N.; Hlushchuk, Y.; Koskinen, M.; Ramkumar, P.; Forss, N.; Kalso, E.; Hari, R. Aberrant temporal and spatial brain activity during rest in patients with chronic pain. Proc. Natl. Acad. Sci. USA 2010, 107, 6493–6497.
- Napadow, V.; LaCount, L.; Park, K.; As-Sanie, S.; Clauw, D.J.; Harris, R.E. Intrinsic brain connectivity in fibromyalgia is associated with chronic pain intensity. Arthritis Rheum. 2010, 62, 2545–2555.
- Cauda, F.; D’Agata, F.; Sacco, K.; Duca, S.; Cocito, D.; Paolasso, I.; Isoardo, G.; Geminiani, G. Altered resting state attentional networks in diabetic neuropathic pain. J. Neurol. Neurosurg. Psychiatry 2010, 81, 806–811.
- Cauda, F.; Sacco, K.; Duca, S.; Cocito, D.; D’Agata, F.; Geminiani, G.C.; Canavero, S. Altered Resting State in Diabetic Neuropathic Pain. PLoS ONE 2009, 4, e4542.
- Schmidt-Wilcke, T.; Leinisch, E.; Gänbauer, S.; Draganski, B.; Bogdahn, U.; Altmeppen, J.; May, A. Affective components and intensity of pain correlate with structural differences in gray matter in chronic back pain patients. Pain 2006, 125, 89–97.
- Apkarian, A.V.; Sosa, Y.; Sonty, S.; Levy, R.M.; Harden, R.N.; Parrish, T.B.; Gitelman, D.R. Chronic Back Pain Is Associated with Decreased Prefrontal and Thalamic Gray Matter Density. J. Neurosci. 2004, 24, 10410–10415.
- Borsook, D.; Upadhyay, J.; Chudler, E.H.; Becerra, L. A Key Role of the Basal Ganglia in Pain and Analgesia—Insights Gained through Human Functional Imaging. Mol. Pain 2010, 6, 1744–8069.
- Neugebauer, V.; Li, W.; Bird, G.C.; Han, J.S. The Amygdala and Persistent Pain. Neuroscience 2004, 10, 221–234.
- Lang, S.; Kroll, A.; Lipinski, S.J.; Wessa, M.; Ridder, S.; Christmann, C.; Schad, L.R.; Flor, H. Context conditioning and extinction in humans: Differential contribution of the hippocampus, amygdala and prefrontal cortex. Eur. J. Neurosci. 2009, 29, 823–832.