1. The Pathophysiology of Back Pain
1.1. Summary of Pathophysiology
Chronic low back pain (cLBP) is complex and affects both the mind and the body and thus needs a multi-disciplinary approach grounded in the bio-psycho-social model
[1][14]. Despite the importance of psycho-social factors, however, prospective studies have shown that these factors predict only 1–3% of future first-time cLBP occurrence
[2][15]. There is no comparable evidence that psycho-social factors initiate cLBP, but instead they act to augment the experience of pain
[3][16]. Evidence for biomechanical and physiological factors as a causative agent of cLBP, however, remain well established in the literature
[3][4][5][16,17,18]. Therefore, to begin exploring what generates the pain in cLBP, these factors must first be outlined. In short, low cLBP begins due to spinal injury or micro-trauma that help further the degeneration of the spine, joints and associated structures. These injuries can either produce serious spinal injury, or much more commonly a type of sub-failure injury, which is defined as trauma done to the spine that is just below the threshold to produce major injury. Specifically, these can originate from three sources: (1) Muscle or ligament strain, (2) Intervertebral disc degeneration, or (3) degenerative joints. These structural changes can cause the following types of pain: myofascial pain, facet joint pain, sacroiliac joint pain, discogenic pain and spinal stenosis
[6][19]. A summary of the pathophysiology of cLBP is illustrated in
Figure 1.
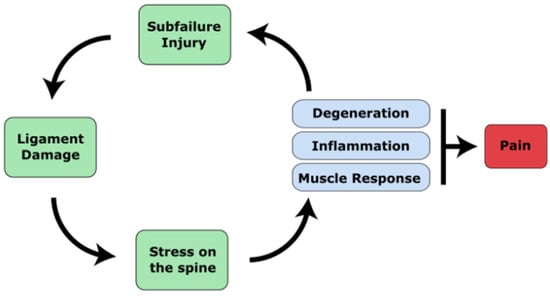
Figure 1. A diagram of the pathophysiology of cLBP. Major trauma or cumulative micro-trauma occurs that damages ligamentous soft tissue. Ligament damage alters the stability of the spine, which increases the risk of poor posture or spinal misalignments that introduce abnormal forces and stresses on the spine. Stress on the spine begins degeneration of the discs and joints, followed by inflammation of the discs as well as compensatory muscular responses. Pain can be caused by disc herniation or bulging that irritates nerves, nerves that grow into the disc, or degenerated facet joints that stimulates nerves. These changes, along with compensatory postures, can increase stress on the spine and further cause micro-trauma, causing pain to persist.
1.2. The Ligamentous Concept
The initiating source of pain from the back is due to abnormal forces acting on the body as a whole and more specifically the intervertebral discs, ligaments and facet joints. The major structural component that passively stabilizes the spine and maintain alignment are the soft tissue ligamentous structures. These include ligaments, joint capsules and intervertebral discs. Damage to these structures predisposes one to cLBP
[4][17]. The major ligaments providing spinal stability are presented in
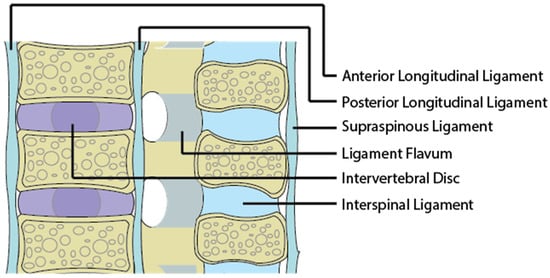
Figure 2.
Figure 2.
An image of a sagittal section of the spine and its associated ligaments.
Damage to the ligaments from trauma or cumulative microtrauma can weaken the spine’s structural capabilities, leading to spinal misalignment. Trauma can occur from events such as a car accident or from repetitive movements in activities such as golfing, gymnastics and cricket
[7][20]. Micro-trauma can also occur from poor postural habits while sitting at an office desk
[4][17]. Abnormal mechanical stresses acting on normal ligaments can itself induce pain if it pinches a nerve. The ligament itself is innervated by neurons that can initiate a painful sensation if disturbed by mechanical or chemical irritation
[8][21]. A misaligned spine can impose high and continuous axial load on the vertebrae, disc and facet joints. Prolonged abnormal stress on ligaments, due to misalignment, initiates degenerative and inflammatory responses that lead to pain.
1.3. Myofascia and Spinal Muscles
Myo-facial pain begins from trauma or repetitive motion injury
[9][22]. This type of pain is characterized by the existence of specific points along the fascia, tendons, or muscles which, if triggered, produces pain
[10][23]. The thoracolumbar fascia (TLF) is one structure that plays an important role in stabilizing the spine. Anatomical studies have shown that the TLF functions to transmit external loads efficiently from the spine to the pelvis, legs and arms
[11][12][24,25]. The TLF also contains many mechanoreceptors that signal information on spinal position
[13][26] and thus the TLF acts similarly to an external layer of structural support to the spine above the spinal ligaments. Other structures that support the spine are the lumbar multifidus and erector spinae. Previous studies have indicated that muscular structure changes from acute to cLBP, showing atrophy, fat infiltration and connective tissue accumulation
[8][14][21,27]. These structural changes are generally explained by compensatory disuse due to changes in movement patterns to safeguard the multifidus from loads
[15][28], pain/fear avoidance
[16][29], or deconditioning
[17][30]. It was reported in one study that fat infiltration into the paraspinal muscle was more relevant than disc degeneration in generating cLBP in women
[18][31]. Another study reported that fat infiltration into the multifidus and erector spinae are highly associated with degenerative discs, which is currently the most commonly reported cause of cLBP
[19][32]. These mechanisms serve to reduce the capability of spinal muscles in providing structural support for the spine, contributing to greater stress on the spine.
1.4. Spinal Muscles and Mechanoreceptors
In addition to the ligaments, spinal muscles also contribute to correct postural alignment of the spine. Once ligament damage and spinal misalignment occur, numerous mechanoreceptors located in the spinal column ligaments
[20][21][33,34], facet capsules
[22][23][24][35,36,37] and disc annulus
[20][33] carry information to the brain. These mechanoreceptors send tactile sensations and position sense to the brain and there is evidence from animal studies in which the stimulation of ligaments
[25][26][38,39], facets and discs
[27][40] results in spinal muscle activation. Specifically, ligament fatigue
[28][41], static flexed posture
[28][41] and cumulative microtrauma
[29][42] have been shown to modulate this form of spinal muscle activation. Under normal circumstances, the mechanoreceptors generate a complex signal which the brain interprets and responds with a muscle response to stabilize the spine. The damaged spine, however, behaves differently. Since mechanoreceptors are imbedded within the discs, ligaments and facet joints, damage from injury as well as degeneration may also lead to damage of these mechanoreceptors
[30][31][43,44]. Damaged mechanoreceptors may send spontaneous signals to the brain regarding body position, a phenomenon referred to as ectopic mechano-sensitivity
[32][33][34][45,46,47]. Unbalanced stresses caused by spinal muscles have been shown to induce strains to ligaments, overload facet joints
[4][17], initiate inflammation of neural tissues
[35][48] and accelerate disc
[36][49] and facet joint degeneration
[37][50]. Therefore, the overall effect of abnormal spinal muscles is to contribute to greater stress on the spine.
1.5. Degenerative Discs and the Inflammatory Response
Intervertebral disc degeneration is an irreversible process characterized by elevated matrix degeneration, nucleus pulposus proteoglycan loss and loss of hydration, destructuration of the disc structure and reduced disc height
[38][39][51,52]. Bending loads, which can be produced by ligament damage and misalignment, can lead to disc prolapse and a cascade of cell-mediated degenerative changes. Bending mechanical stress puts extra pressure along the endplates and annulus of the disc
[40][53], resulting in a bulge or herniation that protrudes outward. If the herniation or bulge causes mechanical compression or distension of the nerve root, dorsal root ganglion, or smaller nerves surrounding the disc, then this will lead to pain via nociceptive neural signals
[41][54]. Further mechanical load can lead to calcification of the end plates, internal disc disruption
[42][43][44][55,56,57] and cell-mediated loss of water content and disc height and is associated with a loss of aggrecan and collagen content within the disc
[45][58]. Structural and material changes of the discs induce ingrowth of nerves and blood vessels within the disc
[46][47][59,60], which then produces painful nerve signals (
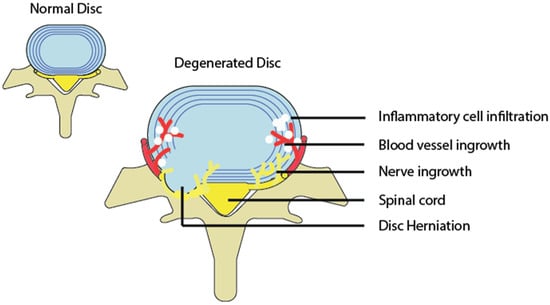
Figure 3).
Figure 3.
A diagram depicting a normal and degenerated disc accompanied by inflammation, as well as nerve and blood vessel growth into the disc.
1.6. Spinal Osteoarthritis and Degenerative Joint Disease
Osteoarthritis of the spine is a type of degenerative joint disease that is best characterized as the breakdown of cartilage in the facet joints. Ligament damage induces abnormal stress onto the facet and is a major cause of joint degeneration
[48][61]. Degeneration of the facets can also occur as a result of degenerated discs, which shift compression loads posteriorly onto the facets. Biomechanical studies support the contribution of mechanical stresses to stimulate degeneration of the facet joints
[49][50][62,63]. Furthermore, evidence from previous work has suggested that facet joint associated pain is caused by mechanical stresses induced by alignment abnormality that leads to degeneration and inflammation of the facet joints
[51][52][53][64,65,66]. The capsule of facet joints is innervated by nociceptive neurons that can be activated by mechanical and chemical stimulation
[54][55][56][57][67,68,69,70]. Direct stimulation of facet joints and ligaments can also stimulate pain
[58][71]. Pain arising from this location is termed Facet Joint syndrome. In neurophysiologic studies using animal models, thirty mechanosensitive units were identified at the lumbar facet joint and twenty-seven were identified in the muscles and tendons near their insertion into the facet
[24][37]. Goldthwaite et al. reported the compression of nerve roots due to a deformation of the facet joint
[59][72]. Since then, nerve root compression by a facet joint has been considered to be one of the causes of low cLBP and sciatica
[60][73].
2. The Transition from Acute to Chronic Pain
Since acute pain is tightly associated with tissue damage, it has an adaptive function as it allows one to focus on caring for the region of the body causing pain. Chronic pain can persist after the tissue damage has healed and therefore does not have an adaptive function. In some cases, chronic pain originates in the absence of any tangible injury, as defined by the international association for the study of pain (IASP)
[61][74]. For pain to go from acute to chronic, the current literature suggests that the morphology and function of the central nervous system itself changes into a pathological state. This transition begins due to acute persistent nociceptive (i.e., pain) stimulation
[62][75]. In peripheral nerves, persistent nociceptive stimulation produces chronic inflammation that leads to reduced pain thresholds for primary neurons, phosphorylation of protein kinases A and C, activation of TRPV1 receptors and the increased production of substance P and CGRP
[63][64][65][76,77,78]. This situation is termed peripheral sensitization and is characterized by an increased sensitivity to afferent nerve stimuli such as heat and touch. This increased sensitivity is also called primary hyperalgesia or primary allodynia if the stimulus was not originally a painful one
[66][79]. Although peripheral sensitization is responsible for the initial sensitization of nociceptors, this mechanism is usually short-lived, reversible and confined to a specific area of the body.
In the spinal cord, persistent nociceptive input causes changes in the dorsal root ganglion (DRG), dorsal horn neurons and glial cells. Gene and protein expression of sodium and TRPV1 channels increase in the DRG, leading to more excitable neurons
[67][80]. NMDA receptors in the dorsal horn neurons also become more excitable, leading to the ‘wind-up’ phenomenon. Wind-up refers to an increase in pain intensity over time; when a given stimulus is delivered repeatedly and clinically, this manifests as allodynia. Prolonged nociceptive transmission to the spinal cord also activates glial cells by increasing neuronal chemokines, neurotransmitters and neuromodulators and produces endogenous danger signals
[65][68][78,81]. Once activated, glial cells release substances into the central nervous system (IL-1, IL-6, TNF, chemokines, prostaglandins, excitatory amino acids, reactive oxygen species and nitric oxide), which has the cumulative effect of enhancing neuronal excitability
[69][70][82,83]. This results in central sensitization, which is a state of reduced thresholds to stimulus associated with the activation of neurons in the spinal cord
[63][76]. This type of sensitization produces secondary hyperalgesia, is long-term, and the pain can spread to other parts of the body not associated with injury.
Pain and temperature are sensed and relayed to the brain via the lateral spinothalamic tract, while crude tactile sensation travels through the anterior spinothalamic tract. Nociceptive signals travel through free nerve endings into the dorsal horn of the spinal cord and progress upward to the thalamus of the brain. The thalamus is responsible for collecting all sensory information and filtering it, only passing information forward upon summation of multiple signals over a threshold. The signal is then sent to multiple cortical areas for interpretation. Disruption in these thalamocortical projections is thought to play an important role in pain perception and also in neuropathic pain. Abnormal oscillatory activity in thalamo–cortico–thalamo loops, termed thalamocortical dysrhythmia, have become a hallmark for chronic conditions such as chronic pain
[71][84]. The transition from acute to chronic pain involves continuous nociceptive input to the brain which alters both brain anatomy and function over time. For example, acute back pain triggers the insular cortex for pain perception, which projects strongly to the nucleus accumbans (NAcc), increasing motivational behaviour in response to pain. As acute pain transitions to chronic pain, the connection between the IS and NAcc decreases, while the connection between the NAcc and medial prefrontal cortex (mPFC) increases. The mPFC is known for processing negative emotions, response conflict and the detection of unfavourable outcomes
[72][85]. Greater pain was also associated with reduced parietal activity corresponding to reduced attentional processing during visual tasks
[73][86]. Other reports have also confirmed that the resting state activity of the brain is distorted in chronic pain conditions
[74][75][76][77][87,88,89,90]. These disruptions of brain activity in chronic pain patients have been shown to produce metabolic changes that lead to degeneration of prefrontal and thalamic grey matter in the brain
[78][79][91,92]. Further brain imaging studies for chronic pain have demonstrated that chronic pain consistently engages the mPFC as well as subcortical limbic areas, including the portions of the dorsal and ventral basal ganglia
[80][93], amygdala
[81][94] and hippocampus
[82][95]. These areas of the brain are responsible for motivation, emotional processing and memory. Therefore, even though different types of chronic pain show a unique activation of brain areas, the transition from acute to chronic pain consistently demonstrates a shift away from purely sensory brain processing towards emotional, motivational and memory processing. These changes may explain observed behaviors such as pain catastrophizing and fear-avoidance.