Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Guo-Ping Lu and Version 2 by Rita Xu.
Carbon-based iron catalysts combining the advantages of iron and carbon material are efficient and sustainable catalysts for green organic synthesis.
- carbon material
- iron
- heterogeneous catalysis
- organic synthesis
1. Introduction
Organic synthesis has made great contributions to the progress of human society, but has also caused serious environmental pollution [1][2][1,2], so there is an urgent demand to introduce green chemistry into this area. One practical tool is the exploration of efficient and selective catalysts for organic synthesis, which can improve synthetic efficiency and reduce production costs and wastes, thereby minimizing the impact on the environment [3][4][3,4].
Iron, as the most productive and cheapest metal [5][6][7][5,6,7], is widely available, biocompatible and non-contaminant. The valence state of iron ranges from −2 to +5, which equips it with flexible redox properties, Lewis acidity and coordination ability [8]. Furthermore, iron-based enzymes are involved in lots of vital and efficient transformations in vivo [9]. All the above mentioned features of iron render a wonderful candidate to replace noble metals, and stand out among other non-precious metals. Therefore, iron-based catalysts meet the requirement of “catalyst economy”, and the development of these catalysts for organic transformations is conductive to the sustainable development of this area [6]. Nevertheless, the high chemical activity of iron also leads to its poor stability, which limits its application possibilities in more catalytic processes [10][11][10,11]. Meanwhile, iron has lower electronegativity and proportion of d orbitals and electrons than noble metals (such as Pd, Ru and Au), so it has poor catalytic activity in hydrogenation and cross-coupling reactions [12][13][12,13].
To solve these issues, ligands or supports are required to stabilize or tune the performance of iron sites [14][15][16][17][14,15,16,17]. Considering that ligands are relatively expensive and non-recyclable, solid supports are undoubtedly more in line with the requirements of green chemistry. Among various supports, carbon materials, with advantages of good biocompatibility, unique microstructure, high specific surface area and excellent physical and chemical properties, are considered excellent supports for iron-based heterogeneous catalysts [18][19][20][21][22][23][24][25][18,19,20,21,22,23,24,25]. Therefore, carbon-supported iron materials combining the advantages of iron and carbon materials are an efficient and sustainable catalyst for organic synthesis. In addition, N-doped carbon anchored iron-single-atom catalysts have similar FeN4 active sites with natural iron-based enzymes in terms of electronic, geometric and chemical structures [26][27][26,27].
Although there are several excellent reviews concerning the organic reactions over heterogenous and homogeneous iron-based catalysts [4][5][6][7][8][28][29][30][31][32][33][34][35][36][37][4,5,6,7,8,28,29,30,31,32,33,34,35,36,37], no attempt focuses on the carbon-based iron catalysts for organic synthesis. In recent years, there have been dozens of organic transformations catalyzed by carbon-based iron materials, including oxidation, reduction, tandem and other reactions. Taking into account the advantages and development trends of nanocarbon-based iron catalysts for organic synthesis, the review on this kind of heterogenous catalysts for organic reactions is of great importance and appeal.
2. General Consideration
2.1. The Introduction Strategies of Iron into Carbon Materials
The catalytic activity of carbon-based iron materials is closely related to their structures, which are controlled by their synthetic strategies. Because numerous reviews on carbon-based metal materials have systematically summarized the controlled fabrication of these materials [38][39][40][38,39,40], this resviearchw mainly focuses on the introduction methods of iron into carbon materials. There are three main steps for the preparation of these catalysts: precursor synthesis, pyrolysis and post-modification (Figure 1). In the precursor synthesis step, iron can be introduced by mechanical mixing, self-assembly and impregnation methods [41][42][43][41,42,43]. Chemical vapor deposition (CVD) can be applied for iron doping during the pyrolysis process [44][45][46][44,45,46]. Impregnation is also an efficient approach for anchoring iron to carbon material in the post-modification step.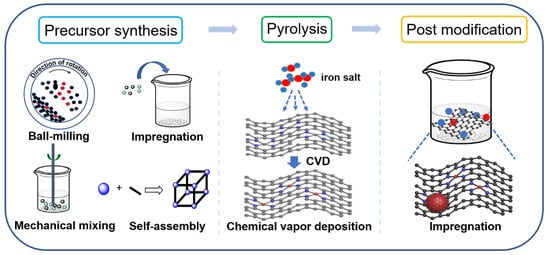
Figure 1. Schematic illustration for the introduction of iron species into carbon supports.
2.1.1. Mechanical Mixing Strategy
Mechanical mixing refers to the direct mixing of iron sources and precursor compounds, without special treatment and requirements on the structure of the precursors. Although this method is easy to be operated, it results in the non-uniformity of carbon particle size and iron distribution [47]. Ball milling has been applied for more even mixing. In 2015, Deng et al. prepared FeN4/GN with highly dispersed FeN4 center by ball milling the mixture of iron phthalocyanine and graphene nanosheets (GNs) [48].2.1.2. Impregnation Strategy
The impregnation strategy is to impregnate the solution of iron source with carbon materials or carbon precursors, with the removal of residual solvent after impregnation equilibration then leading to the introduction of iron species into carbon materials or carbon precursors [49]. In general, the impregnation method has better mixing efficiency than mechanical mixing, resulting in a more uniform Fe doping. However, the impregnation method still cannot effectively solve the problem on the inhomogeneity of carbon particle size and iron distribution. For example, ethyl cellulose was dissolved in ethanol and mixed with the aqueous solution of Fe(NO3)3·9H2O, and then melamine and zinc nitrate were added as nitrogen source and pore former, respectively (Figure 2a) [50]. However, the size and distribution of carbon and iron particles in Fe@CN-Zn are not uniform, which can be observed from the results of TEM (Figure 2b) and SEM (Figure 2c).
Figure 2. (a) Schematic illustration of the synthesis of Fe@CN-Zn. (b) TEM and (c) SEM images of Fe@CN-Zn. Reprinted with permission from Ref. [50]. Copyright 2021, Elsevier.
2.1.3. Self-Assembly Strategy
The introduction of iron can also be achieved through the self-assembly of iron salts with certain organic ligands to form ordered 3D porous metal-organic frameworks (MOFs), which can be divided into two types: Bimetallic MOFs and Fe-based MOFs. Compared to other methods, nanocarbon-based iron catalysts prepared from self-assembly precursors have advantages of ordered porous structure, more uniform carbon size and iron distribution [51][52][53][51,52,53]. However, this approach has special requirements for organic ligands, which should be able to coordinate with iron ions to form MOFs. As a bimetallic MOF strategy example, Fe-ZIF (ZIF represents zeolitic imidazolate framework) was prepared via the self-assembly of iron salts and zinc nitrates with 2-methylimidazoles (Figure 3a) [43]. An N-doped carbon-based iron catalyst was synthesized by the pyrolysis of Fe-ZIF. As a representative example of Fe-based MOF strategy, MIL-101(Fe) was prepared by self-assembly of 1,4-terephthalic acid with iron nitrate hexahydrate, which was grinded with melamine and then calcinated to obtain Fe/Fe2O3@NnPC-T-x catalyst (n represents the ratio of MIL-101 (Fe) and melamine, T represents calcination temperature and x represents calcination pyrolysis time) (Figure 3b) [54].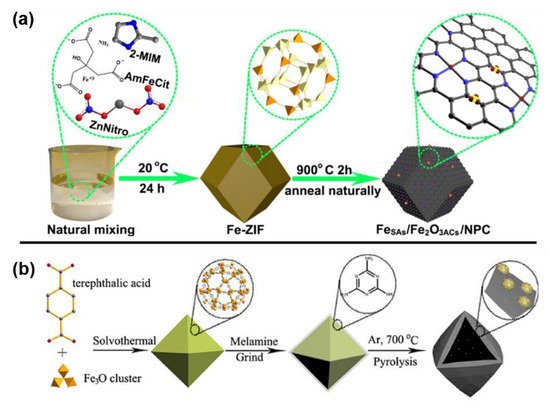
Figure 3. (a) The preparation of FeSAs/Fe2O3ACs/NPC from bimetallic MOF strategy. Reprinted with permission from Ref. [43]. Copyright 2020, American Chemical Society. (b) The preparation of Fe/Fe2O3@NnPC-700-x from Fe-based MOF strategy. Reprinted with permission from Ref. [54]. Copyright 2019, Wiley.
2.1.4. Chemical Vapor Deposition (CVD)
Chemical vapor deposition (CVD) refers to a vapor phase self-assembly carbonization process at high temperatures, which can effectively control the chemical composition, morphology, crystal structure and grain size of the membrane layer by adjusting the deposition parameters [55]. This method can make iron species in full contact with the carbon materials during the carbonization process, constructing a unique active iron center, and finally obtaining a catalyst for carbon-loaded iron with excellent catalytic activity. In 2021, Jia et al. prepared a Fe-N-C catalyst containing dense FeN4 sites by flowing iron vapor through the Zn-N-C substrate at 750 °C (Figure 4) [56].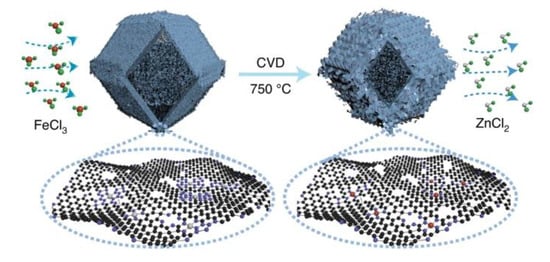
Figure 4. Preparation of FeNC-CVD-750 via CVD strategy. Reprinted with permission from Ref. [56]. Copyright 2021, Springer Nature.
2.2. The Structure and Activity Relationship (SAR)
2.2.1. Nature Properties of the Carbon Materials
In comparison of metal oxide supports, carbon materials have unique porous structure, and are easier to be doped with heteroatoms, which can adjust their Lewis acid-base sites and defect the degree and electronic state of Fe sites, facilitating the formation of stable and active iron catalytic sites. Therefore, iron-doped carbon materials exhibit superior performance than metal oxide-supported iron catalysts in various transformations. For example, Beller’s group have prepared two carbon-supported iron catalysts: Fe-phenanthroline/C-800 [57] and Fe-L1@EGO-900 [41], which exhibit good catalytic activity for the reduction of nitroarenes and acceptorless dehydrogenation reactions, respectively. The two catalysts were prepared by calcinating the mixture of iron precursor, 1,10-phenanthroline ligand and carbon support. The difference is that carbon powder was used as carbon support in Fe-phenanthroline/C-800, while exfoliated graphene oxide (EGO) was applied as carbon support in Fe-L1@EGO-900. However, the catalytic efficiency of other heterogeneous iron catalysts is much lower, or even with no catalytic effect when the carbon material is replaced with other carriers, such as Al2O3, SiO2, CeO2 and TiO2. Overall, the porous structure and defects of carbon supports are favorable for the interaction between substrates with active sites, as well as the exposure of active sites in the catalysts. The Lewis acid-base sites of carbon supports can improve the adsorption and activation of substrates.2.2.2. Iron Sites in Carbon Materials
Generally, there are two types of iron sites in carbon materials: single atomic iron sites and iron nanoparticles (NPs) [58][59][58,59]. The identification of these two iron sites can be achieved by XRD, TEM, XAFS and HAADF-STEM [60][61][62][60,61,62]. Single atomic iron sites and iron nanoparticles (NPs) can usually play a synergistic catalytic role by exerting their respective catalytic advantages in different steps of one reaction [58][60][63][58,60,63]. Although some control experiments have been designed to verify the synergistic catalytic effect between these two sites, the research on this aspect is still in its infancy. For example, Yang et al. designed a series of control experiment to confirm the synergistic catalysis on FeNx sites (single atomic Fe sites) and Fe NPs for the fabrication of quinolones and quinazolinones (Figure 5a) [58]. Because acid etching can remove Fe NPs selectively, while SCN− can specifically poison FeNx sites, the catalytic effects of FeNx and Fe NPs in the reaction can be clarified. In addition, the reaction principles of Fe(II)Pc and nano-Fe powder are similar to that of FeNx sites and Fe NPs, which are also applied to detect the role of FeNx and Fe NPs during the catalytic process. The coupling reaction of amine and aldehyde and the oxidative dehydrogenation of intermediate I are the two steps involved in the oxidative coupling reaction (Figure 5a). As shown in Figure 5a, FeNx sites are more favorable for the formation of intermediate I, while Fe NPs are more conducive to the further dehydrogenation of intermediates I to produce quinolones and quinazolinones. Thus, FeNx sites and Fe NPs play a synergistic catalytic role in this oxidative coupling reaction.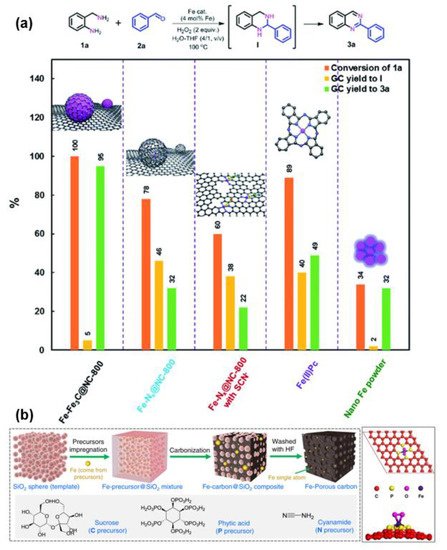
2.2.3. The Construction of SAR
The construction of SAR between carbon-based iron catalysts and organic reactions are mainly explored by control experiments, reaction kinetics studies and density functional theory (DFT). Control experiments are performed to determine possible reaction intermediates and reaction pathways by changing the reaction conditions or substrates appropriately, or by adding additives. As shown in Scheme 1, the role of Fe-Fe3C@NC-800 and H2O2 in the reaction is investigated by control experiments. Based on the results of Scheme 1, it can be concluded that Fe-Fe3C@NC-800 is necessary for both the coupling and dehydrogenation process, while H2O2 is favorable for the dehydrogenation process [58].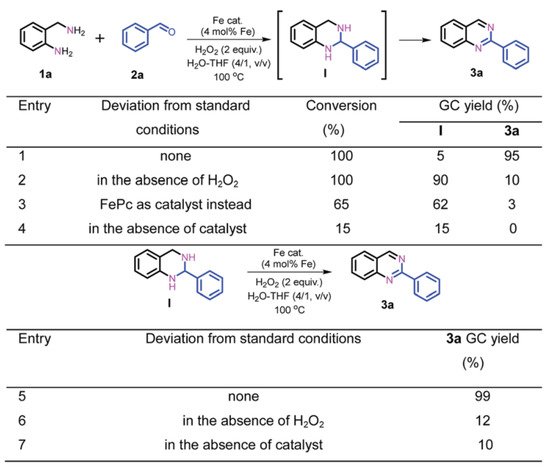
Scheme 1.
The role of Fe-Fe
3
C@NC-800 and H
2
O
2
in mechanism exploration. Reprinted with permission from Ref. [58]. Copyright 2019, Royal Society of Chemistry.
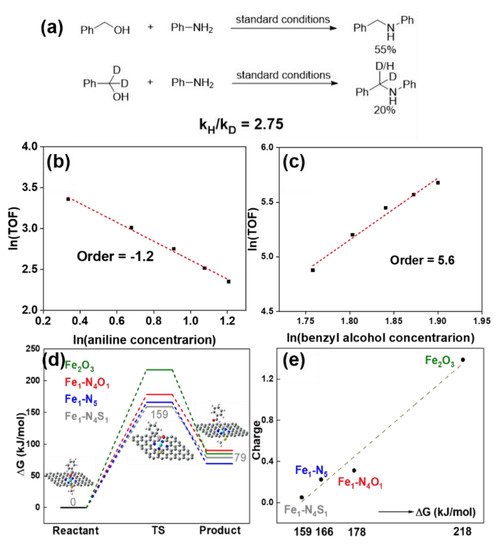
Figure 6. (a) The H/D kinetic isotope effect (KIE) experiment results. Plots of reaction orders of (b) aniline and (c) benzyl alcohol. (d) The energy barriers for Fe2O3, Fe1-N4O1, Fe1-N5 and Fe1-N4S1 sites. (e) The relationship between positive charge density of Fe sites and energy barriers. Reprinted with permission from Ref. [66]. Copyright 2021, Royal Society of Chemistry.