New crystals of PbF(IO3) polytype modification are synthesized hydrothermally and demonstrate strong SHG optical response. They are phase-matchable at the fundamental wavelength of 1064 nm. The crystal structure was solved in two space groups, orthorhombic C2ma and monoclinic Pn, of which monoclinic is true and is described with a twinning by mirror plane introduced in structural refinements taken into account. Orthorhombic symmetry was used in comparison with the related structures and deviation close similarity in the selected suggested family MX(IO3), M = Bi, Ba, Pb, X= O, F, (OH) with series of members. These compounds were also characterizing as similar to Aurivillius phases with fluorite-like layer and perovskite-like layer substituted by (IO3) groups. The optical nonlinearity of the iodates of the Aurivillius family and structurally related iodates is determined by the polar orientation of the iodate groups, which make an overwhelming contribution to the optical nonlinearity. From crystal chemistry point of view, the heavy atoms in these structures are located in the second cation environments in relation to the iodate groups and indirectly affect the nonlinearity. In particular, large Ba-cations without single electron pairs provoke a symmetric variant of the Aurivillius type structure, in contrast to the acentric Bi3+ and Pb2+ cations known in polar iodates with strong second-order optical nonlinearity. There is wide diversity in the extended series of related compounds which includes variation of fluorite-like layers (single or double), perovskite-like layers presented by octahedral or more complicate polyhedral, or by IO3 (BrO3) groups, or by Cl-atoms, or by NH4-groups. This allows the development of future search for new promising phases.
- compound
1. SHG, Orthorhombic Model, Ambient Conditions Data
The SHG output from 100 mcm PbF(IO3) powder is approximately 2 times larger in relation to analogous KDP (KH2PO4) powder, which is close to other metal iodate fluorides (Figure 1).
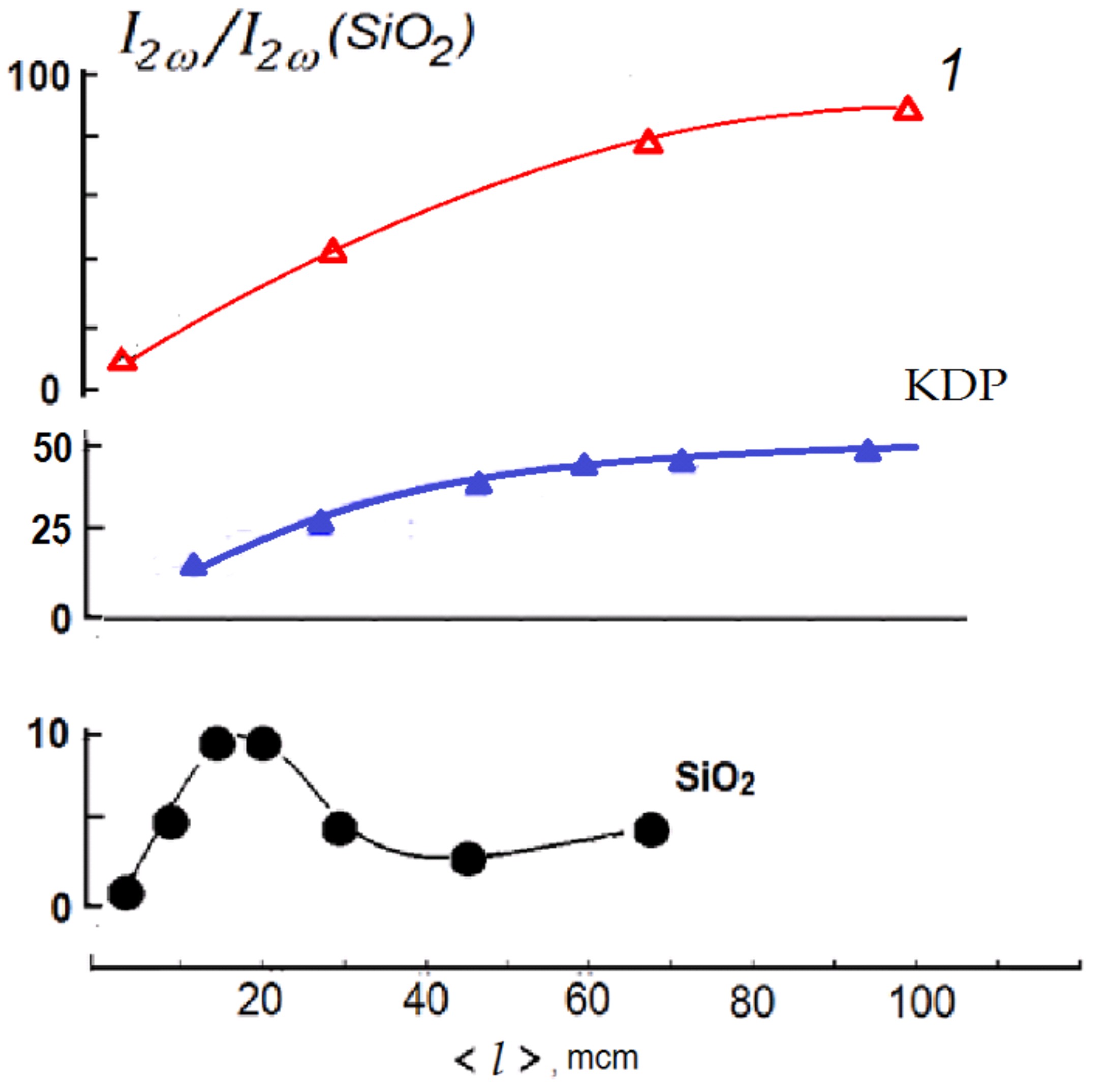
Figure 1. Powder SHG response of two experiments of PbF(IO3) shown as 1 in comparison with a-quartz and KH2PO4 in dependence on average grain size in the powders.
2. Monoclinic Model, Low-Temperature Data
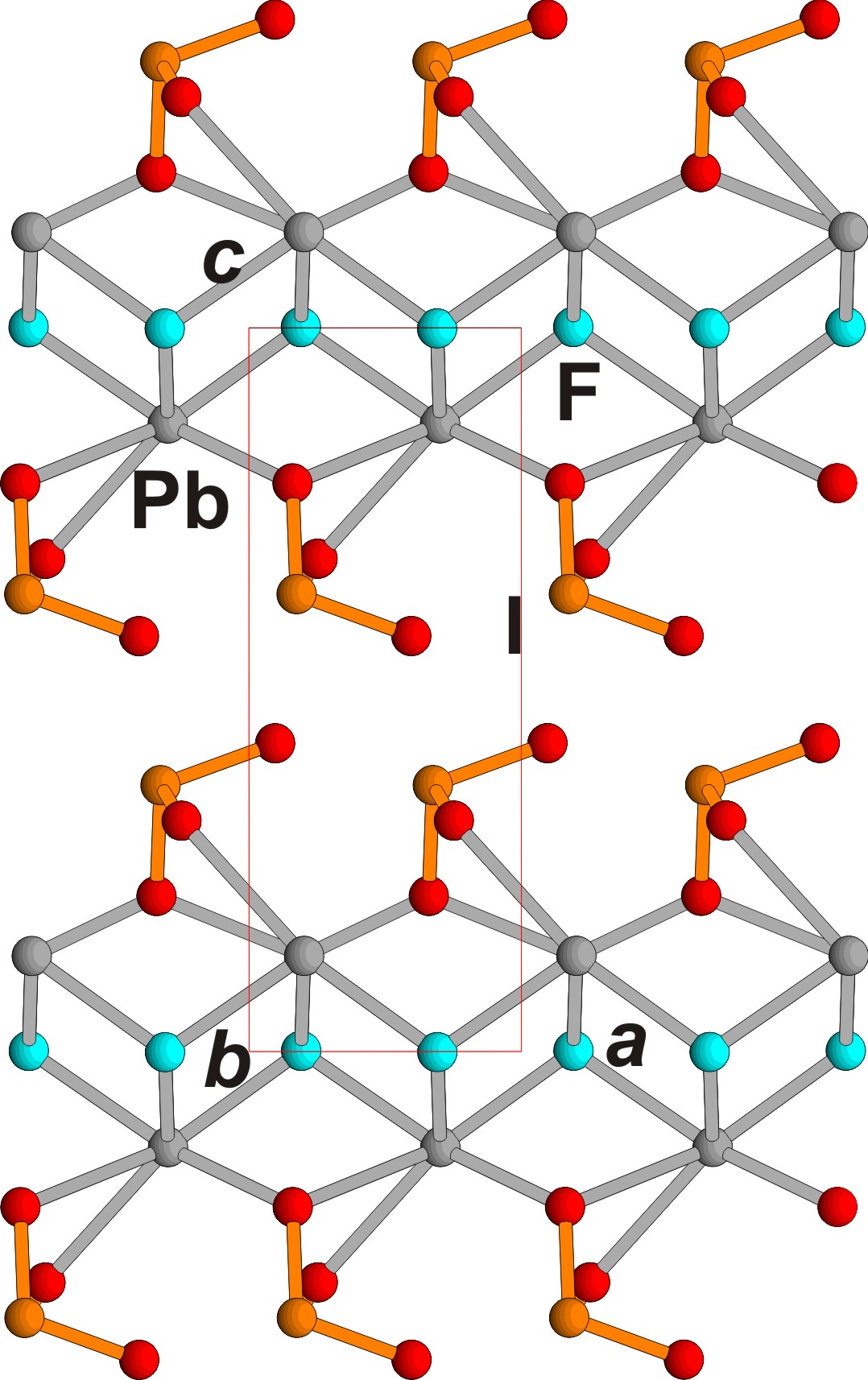
Figure 3.
PbF(IO
3
) crystal structure,
ac
projection. Pb, I, F and O ions are shown in grey, orange, blue and red respectively.
The crystallographic data, atomic coordinates, and selected bonds are presented in Table 1. CCDC (ICSD) 2212814 and 2212815 contain, accordingly, the crystallographic data for the orthorhombic average structure and monoclinic true structure, refined with a twin component. These data can be obtained free of charge via , Table S1–S4. CCDC (ICSD) 2212814 and 2212815 contain, accordingly, the crystallographic data for the orthorhombic average structure and monoclinic true structure, refined with a twin component. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif (accessed on 13 October 2022). All the illustrations were produced using the ATOMS [5] and CORELDRAW programs. (accessed on 13 October 2022). All the illustrations were produced using the ATOMS [12] and CORELDRAW programs.Orthorhombic symmetry for new compound was used in comparison with the related structures and deviation close similarity in the selected suggested family MX(IO3), M = Bi, Ba, Pb, X= O, F, (OH) with series of members: orthorhombic PbF(IO3), monoclinic PbF(IO3), BiO(IO3), Ba(OH)(IO3) and BaF(IO3). All of them have polytypic nature with sheets composed of central pseudo-tetragonal anion-centered layer (MX) similar to layer in PbO lithargite structure and attached from its both sides (IO3) groups. The central layer has also description as fluorite-like layer. These compounds were also characterizing as similar to Aurivillius phases with fluorite-like layer and perovskite-like layer substituted by (IO3) groups.
TheOur new crystals demonstrate disorder presented as twinning. It is proved that in theour experiment researcherswe obtained the simplest structure in the whole polytype family which has the smallest unit cell and the lowest symmetry in contrast with previously determined polytype and other members of the family. ResearchersLet us analyzed the reason of formation of different polytypes. Despite the fact that new modification of PbF(IO3) was synthesized under hydrothermal conditions, and the previously known one was obtained during unconventional low-temperature solid-phase technology, the conditions for obtaining both phases are similar. Both syntheses proceeded at a temperature of 270°C in autoclaves of approximately equal capacity. HIO3 and NH4NO3, which were used to obtain the previous modification, decompose at a temperature of about 200°C to form water, thus the reaction proceeded in an aqueous solution. The main difference in the formation of two polytypes is the reaction rate and the amount of solvent. The synthesis of the phase described in this article proceeds with a large amount of solvent at a lower reaction rate, which contributed to the formation of simpler polytype structure. The growth conditions of previously obtained PbF(IO3) modification favor to synthesis of the most complicate polytype in the family.
The optical nonlinearity of the iodates of the Aurivillius family and structurally related iodates is determined by the polar orientation of the iodate groups, which make an overwhelming contribution to the optical nonlinearity. From crystal chemistry point of view, the heavy atoms in these structures are located in the second cation environments in relation to the iodate groups and indirectly affect the nonlinearity. In particular, large Ba-cations without single electron pairs provoke a symmetric variant of the Aurivillius type structure, in contrast to the acentric Bi3+ and Pb2+ cations known in polar iodates with strong second-order optical nonlinearity.
There is wide diversity in the extended series of related compounds which includes variation of fluorite-like layers (single or double), perovskite-like layers presented by octahedral or more complicate polyhedral, or by IO3 (BrO3) groups, or by Cl-atoms, or by NH4-groups. This allows the development of future search for new promising phases.