1. Following a Self-Catastrophic Path—Missing the Balance
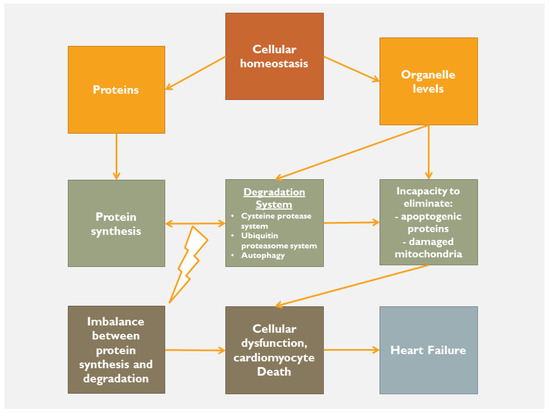
Following an acute index event, the body, as a whole, tries to retain its homeostatic status. If the cause is of minimal aggressiveness, then the homeostatic status remains within normality by using low adaptation mechanisms. However, in the case of a major index event, the body tries to maintain homeostatic status by any means in order to limit the cause, to heal, resolve and ultimately to repair the tissues’ structure and function. In this respect, when a severe disturbance of homeostasis occurs, then the inflammatory process is activated as the acute-intermediaterestore phase, followed, in case of failure of the above-described sequence, by the chronic phase. Regardless of the cause of a sterile inflammation, there is tissue damage and consequently a release of intracellular (nuclear and/or cytosolic proteins, etc.) and extracellular (hyaluronic acid, fibronectin, etc.) products. The release of these proteins activates a series of injury-associated molecular pathways through cardiac receptor signaling. At the beginning, release of inflammatory cytokines, neutrophil aggregation and activation, release of proteases and ROS production occur. Failure of this initial reaction to restore tissue integrity activates a forward step of inflammation, in which the toll and nucleotide binding and oligomerization domain (NOD)-like receptors (NLRs) are involved with further accumulation and activation of pro-inflammatory mediators. At this crucial phase, it is very important to maintain equilibrium between protein degradation (cysteine-protease system, ubiquitin proteasome, autophagy, etc.) and protein synthesis. If this equilibrium fails, apoptogenic mediators, misfolded proteins and damaged mitochondria lead to the phase of chronic inflammation (
Figure 1). The NLRs, joined by caspase-activity complexes, form the inflammasome that further stimulates the production of IL-1b and IL-18 that affect left ventricular systolic function, alter mitochondrial function and decrease sympathetic activity
[1][2][24,25].
Figure 1.
Deranged homeostasis leading to heart failure.
The role of NLRP3 inflammasome (NLR family, pyrin domain-containing 3) in heart failure is well documented
[3][4][5][26,27,28]. NLRP3 inflammasome sets off the maturation of proinflammatory cytokines (IL-1β and IL-18) to initiate the inflammatory response and plays a key role in modulating chronic inflammation, altering the physiological adaptation of cardiomyocyte and leading to heart failure progression
[3][26]. Recent data showed that two other inflammasomes seem to be involved in the inflammatory process in failing hearts. Inflammasome protein absent in melanoma 2 (AIM2) and NLR family CARD domain-containing protein 4 (NLRC4) have been found to be over-expressed and activated in human-heart tissues as well in vivo animal models. These two other inflammasomes may contribute to the chronic inflammation in heart failure and also a therapeutic target
[4][27]. The inflammasome also defines the interplay between innate and adaptive responses, paving the way toward the development of heart failure. Furthermore, the involvement of the immune process (effect of T and B cells) promotes chronicity according to the self-antigen hypothesis, the production of autoantibodies and tissue fibrosis, suggesting a role for autoimmune mechanisms
[6][7][22,29]. This self-protection/elimination process integrates the endogenous inducers, cell-, tissue-, plasma- and extracellular matrixderived signals and might develop in an uncontrolled manner. Any injured myocardial cells can maintain a basal, stressed, apoptotic or necrotic state. If the amount of injured tissue is enormous and overpasses the homeostatic capacity to restore cell-tissue normality, then the detrimental chronic inflammatory phase develops
[8][30]. On the other hand, the successful restoration of homeostasis prevents the harmful effect of chronic inflammation
[9][10][31,32].
2. Homeostatic Mechanisms
To achieve homeostasis, a balanced activity between protein synthesis-degradation and organelle capacity to eliminate apoptogenic proteins and damaged mitochondria should be activated and well-functioning. If this is not the case, then the cardiomyocyte death along with extra-cellular cardiac matrix dysregulation, lead to myocardial cellular dysfunction and ultimately to heart failure (
Figure 14). In other words, the body tries to protect itself from itself. Indeed, when mitochondrial morphology and function are disturbed (lack of fission, fusion and hence mitophagy), mitochondrial DNA is released into cytosol, and along with the misfolded proteins and the activation of the mitochondria-associated endoplasmic reticulum membranes (MAMs), promotes the enhancement of a self-destruction process, that might involve the entire body
[11][12][33,34]. In case of a cardiac harmful event, there is an activation of danger-associated molecular patterns (DAMP) released by the nucleus (e.g., DNA, RNA), the mitochondria (e.g., DNA) and the cytosol (e.g., RNA). In this respect, regardless of the initial triggering event (pressure overload, volume overload, myocardial infarction, etc.), there is an activation of an inflammatory process associated with the harmful release of cell proteins along with the activation of the aforementioned self-elimination/protection mechanism. Thus, if there is an imbalance of this sequel, then the chronic inflammation is switched on, and in case of an uncontrolled process, heart failure develops. In other words, it seems that if the homeostatic mechanism (degradation system, autophagy, etc.) is successful, inflammation is limited. On the other hand, if the homeostatic protective mechanism cannot control and limit the harmful events, the self-catastrophic pathway promotes cardiomyocyte death and hence heart failure. The inevitable question that arises is whether the cause of heart failure is inflammation per se or the incapacity of the homeostatic protective mechanisms.
Damaged and un-repaired mitochondria are the source of reactive oxygen species, and along with mitochondrial DNA release, generate proinflammatory cytokines and the activation of inflammasome, promoting inflammation chronicity. This leads to an increase of the rate and amount of myocardial cell death and hence to the development of heart failure. Although the role of inflammasome (and its subfamilies) is not very well understood, it appears that its formation and activation have dual contradictory roles. The first one is to eliminate the ‘enemy’ and restore the normal anatomy and function of the tissue, while the second one, under certain circumstances, could be harmful by distorting the normal activity, which is to avoid chronic inflammation and to promote the protective mechanisms of homeostasis; in other words, to recognize the released material as foreign and to attack these unrecognized substances in order to ‘protect’ the cell and consequently the normal anatomy and function of the tissue
[13][14][35,36].
It should be stressed that cardiomyocyte homeostasis as described above is different from heart (organ) and body homeostasis. The heart as an organ tries to adapt to stressors and noxious agents mediated by inflammation and redox disorders with an effort to maintain its function in the human body.
3. Organelle Communication
The normal function of a cell depends mainly on the structural functional integrity of its constituents, the organelles. The endoplasmic reticulum (ER) is an organelle that regulates important intracellular function, including protein synthesis, calcium transportation, etc. In the case of an index event, the ER is stressed and tries to maintain normality through homeostasis. In fact, ER-associated degradation, the unfolded protein response, reticulophagy, proteostasis, autophagy, etc., are activated in order to maintain normality
[15][16][17][37,38,39]. In addition, there is communication with the other organelles, lysosomes, mitochondria, plasma membrane, etc., thus facilitating the normal functions of the cell, including lipid metabolism
[18][40], calcium homeostasis
[15][19][37,41], ion exchange
[18][40], etc. However, if the index event surpasses the capacity of the cell to retain homeostasis or if ER homeostatic properties are impaired, then the cell-defending mechanisms fail, thus leading to a possible harmful path
[20][21][22][42,43,44].
Although there is vast communication among the organelles, it seems that the most important one is between the ER and mitochondria
[23][24][45,46]. Indeed, these two organelles form the ER-mitochondria contacts (ERMCs)
[25][47], constituted by both lipid and protein complexes
[26][48]. Studies have demonstrated that ERMCs are involved in the progression of several cardiovascular diseases
[18][27][28][29][30][31][40,49,50,51,52,53], because they are involved in several biological processes, such as calcium homeostasis, apoptosis, autophagy, protein synthesis and folding, inflammation etc.
[32][33][34][35][36][37][38][39][54,55,56,57,58,59,60,61]. After an index event, misfolded proteins are accumulated in the ER promoting the activation of the unfolded protein response in order to maintain proteostasis. In the case of failure of the misfolded protein repair, or of a large amount of accumulated unfolded proteins, a vicious circle begins
[40][41][62,63]. This vicious circle is characterized by the loss of homeostatic capacity, promoting apoptosis. However, ER activation facilitates steroid synthesis, ER stress, phospholipid metabolism in mitochondria, autophagy and apoptosis
[41][63], and under certain circumstances can increase transcription-factor expression (ATF) 6 and 4 and promote apoptosis either alone or in cooperation with mitochondria
[42][43][44][64,65,66]. A self-catastrophic sequence thus begins. Indeed, when the collaboration between these two organelles is impaired, a progression to advanced heart failure may occur
[45][46][67,68]. In fact, it has been stated that uncontrolled ER stress provokes distortion of myocardial architecture, alteration of mitochondrial metabolism and function, leading to an energy deficiency, along with a reduction of calcium transfer and consequently impairment of cardiac contractility and relaxation, hence heart failure
[47][48][69,70].
4. Targeting Inflammation, Oxidative Stress and Mitochondrial Dysfunction
Regardless of whether the inflammation is the cause or the consequence of heart failure, it remains an important factor and a potential therapeutic target
[49][71]. Although, several studies have been conducted in order to investigate the role of anti-inflammatory therapies, the results have hitherto been poor or controversial
[50][72]. Notably, anti-cytokine therapies were tested in the ATTACH and RENEWAL studies with poor results
[51][52][73,74]. On the other hand, the CANTOS trial has shown that the inhibition of IL-1b with canakinumab was followed by a significant trend for a dose-dependent reduction in the incidence of the composite endpoint of hospitalization for heart failure and heart failure-related mortality
[53][75]. However, this was not the case in other studies, showing that after IL-1b inhibition with canakinumab, substantial residual inflammatory risk remained, related to both IL-18 and IL-6
[54][76]. Other studies based on anti-inflammatory therapies have been published
[55][56][57][77,78,79], among which those using either immunomodulation
[58][80] or anti-inflammatory drugs
[59][60][61][81,82,83], showing overall poor results. The same was true when N-terminal pro-B-type natriuretic peptide (NT-pro BNP) or high-sensitivity C-reactive protein (hs-CRP) were used as endpoints
[62][63][84,85].
These data support the need for a better understanding of the inflammatory process. As it has been pointed out, important inflammatory mediators are released after the activation of the inflammasome, suggesting that the inflammasome could be a therapeutic target. Since the inflammasome is part of homeostatic mechanism, one could speculate that homeostatic controlled response is the master key to investigate and target.