Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Rita Xu and Version 1 by ShaoQiang Wei.
Pyroptosis is an active and ordered form of programmed cell death. The signaling pathways of pyroptosis are mainly divided into canonical pathways mediated by caspase-1 and noncanonical pathways mediated by caspase-11. Cell pyroptosis is characterized by the activation of inflammatory caspases (mainly caspase-1, 4, 5, 11) and cleavage of various members of the Gasdermin family to form membrane perforation components, leading to cell membrane rupture, inflammatory mediators release, and cell death. Moderate pyroptosis is an innate immune response that fights against infection and plays an important role in the occurrence and development of the normal function of the immune system.
- pyroptosis
- inflammatory caspase
- inflammasome
- pyroptosis blockers
1. Introduction
The modes of cell death include programmed and non-programmed cell death. Programmed cell death refers to the programmed process of cell death in order to maintain the stability of the internal environment after cells receive a certain signal or are stimulated by certain factors. Apoptosis, autophagy, programmed necrosis, and pyroptosis are the manifestations of programmed death. Thereinto, pyroptosis is a newly discovered form of programmed cell death, which was firstly discovered by Zychlinsky et al. in macrophages infected with Shigella flexneri, because its morphological characteristics were significantly different from apoptosis [1]. Later, Cookson et al. found that it is dependent on the activity of caspase-1 and different from caspase-3-activity-dependent apoptosis, defined this form of cell death as caspase-1-dependent cell death for the first time, and proposed the concept of pyroptosis for the first time [2]. Due to the dependency on inflammatory caspase-1, pyroptosis primarily refers to inflammatory cell death, which is obviously different from apoptosis and necrosis. Meanwhile, with the deepening of research on pyroptosis, it is found that pyroptosis plays an important role in the occurrence and development of many diseases.
Pyroptosis is an important innate immune response that is critical in fighting infection. It is characterized by the expansion of cells until the cell membrane ruptures, resulting in the release of intracellular contents and activating the body’s strong inflammatory response. IL-1β and IL-18 released by pyroptosis cells are endogenous immune factors that cause fever, stimulate the activation of immune cells, and promote lymphocyte proliferation and the secretion of antibodies, while excessive pyroptosis leads to the uncontrolled release of IL-1β and IL-18, which will lead to a wide range of inflammatory reactions and immune diseases [3].
Pyroptosis is a double-edged sword; moderate pyroptosis contributes to the stability of the intracellular environment and plays an important role in fighting infection through eliminating bacteria to protect the host, but excessive pyroptosis not only leads to immune desensitization, but also causes life-threatening diseases such as sepsis, cytokine release syndrome (CRS), severe inflammation, and tissue damage [4,5,6,7][4][5][6][7]. Thus, excessive pyroptosis warrants serious attention during disease treatment. In order to decrease the adverse effects of excessive pyroptosis, the potential pyroptotic blockers have been extensively explored recently, which can keep pyroptosis within a reasonable range through inhibiting or regulating the pathway of pyroptosis. Understanding the occurrence and regulatory mechanism of pyroptosis and identifying potential inhibitory drugs may provide a new direction for further research on pyroptosis.
2. Characteristics of Pyroptosis
2.1. Morphological Characteristics of Pyroptosis
Pyroptosis has some characteristics of necrosis and apoptosis in morphology, but it is different from apoptosis and necrosis. When cells undergo pyroptosis, cytoplasm membrane is ruptured and forms between 1–2 nm in diameter holes, resulting in the release of intracellular substances, the outflow of potassium ions, and cell swelling [8]. The substances stimulate the body’s immune response, recruits more inflammatory cells, and expands the inflammatory response. In this process, the nuclei gradually becomes round and nuclear condensation occurs, and the chromatin DNA breaks and degrades randomly [9,10,11][9][10][11].
2.2. Molecular Characteristics of Pyroptosis
2.2.1. Inflammatory Caspases
Caspases were first discovered from nematodes in 1993 and are mainly involved in cell apoptosis and inflammatory reaction [12]. At present, 15 caspase family members have been found in mammals, including 13 caspases in humans and 11 caspases in mice [13]. Caspases are evolutionally conserved intracellular proteases with homology and similar structural features. Their active sites contain cysteine residues that can specifically cleave the peptide bond behind the aspartic acid residues of target proteins and are known as aspartic acid-specific cysteine proteolytic enzymes [14]. Caspase has been proved to be an essential protease for the developmental death of biological somatic cells. In normal cells, caspase usually exists in an inactive proenzyme state (pro-caspase) that can become active caspases after hydrolysis of amino acid sequences, thereby cutting relevant substrates, leading to the activation, inactivation, repositioning, or remodeling of substrates to play its role [15].
According to the differences in structure and function, caspases can be divided into apoptotic and inflammatory classes. Apoptotic caspases were related to apoptosis including caspase-2/3/6/7/8/9/10 and represented by caspase-3, while inflammatory caspases mediate inflammatory reaction and pyroptosis including caspase-1/4/5/11/12/13/14 [16,17][16][17]. Among inflammatory caspases, mouse caspase-1/11 or human caspase-1/4/5 are key proteins mediating pyroptosis pathway. Caspase-1, the first identified member of the caspase family, is responsible for cleaving pro-interleukin-1beta (pro-IL-1β). In addition, caspase-1 can cleave pro-interleukin-18 (pro-IL-18) into the mature form of IL-18 to play an immunomodulatory function [18,19,20,21,22,23][18][19][20][21][22][23]. Caspase-11 exists only in rodents, and mouse caspase-11 and human caspase-4/-5 are evolutionarily homologous genes [3]. Functionally, both human caspase-4/-5 and mouse caspase-11 can recognize LPS in cells ·.
In addition to executioner caspases, activation of almost all caspases requires removal of the interdomain linker (IDL) and the prodomain [24,25,26,27,28,29][24][25][26][27][28][29]. The prodomain has multiple functions: first, the removal of the prodomain is an important step in caspase activation; second, the prodomain plays an important role in promoting caspase; third, the predomain has a stabilizing effect, and removal of the predomain would inactivate the caspase [21,26,30,31,32,33,34,35,36,37,38][21][26][30][31][32][33][34][35][36][37][38]. As shown in Figure 1, pro-caspase-1 also shares these common sequence features, with its N-terminal prodomain consisting of CARD (residues 1–95), a CARD domain adaptor (CDL) to the caspase domain, and an IDL between the p20 and p10 subunits. The catalytic C285 cysteine residue is located within the P20 region; for instance, self-proteolytic cleavage occurs at three aspartate residues (D103, D119, D297, and D316), releasing the prodomain (CARD, CDL, IDL), which is a key step in human caspase-1 activation [21,39][21][39]. There are two models for caspase-1 activation. The first model proposes that pro-caspase-1 dimerization is followed by self-proteolytic cleavage, while the second model has self-proteolytic cleavage before dimerization [40]. Recent results are more consistent with the previous dimerization–autoproteolytic model [32,41][32][41]. Recruitment of pro-caspase-1 to the inflammasome platform contributes to its activation. Various reports have indicated that caspases are activated by substrate-induced or adjacent-induced oligomerization followed by autoproteolytic cleavage [21,28,29,31,32,41,42,43,44,45,46,47][21][28][29][31][32][41][42][43][44][45][46][47].
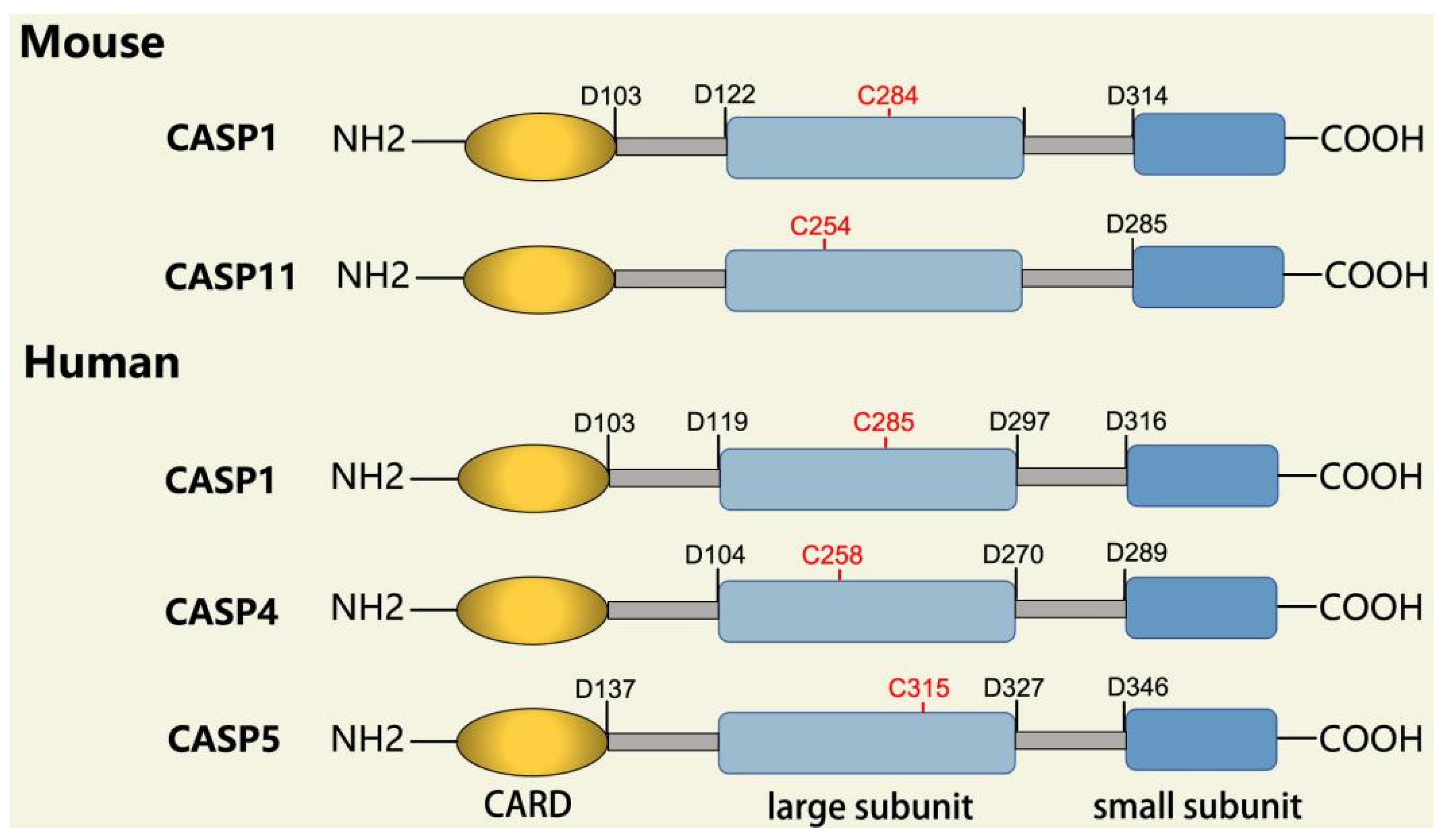
Figure 1. Domain structure of mouse (caspases-1, -11) and human (caspases-1, -4, -5) inflammatory caspases. A CDL connects N-terminal CARD to the protease domain that is composed of a large subunit (p20) and a small subunit (p10) separated by an IDL. Each caspase contains some autocleavage sites (black site) within the linker sequences. The catalytic cysteine (red site) is located within the large protease subunit, while the dimerization interface is within the small subunit.
Caspase-11 is essentially an endogenous receptor of LPS. For caspase-11 activation, LPS directly binds to the CARD domain of caspase-11 via its lipid A tail [48]. In the case of Gram-negative bacterial infections, the hexacylated lipid A moieties of LPS bind directly to the caspase-11 CARD domain, resulting in oligomerization and adjacent induced activation of caspase-11, which may be facilitated due to the polymeric tendencies of LPS. Cytoplasmic pentacylated and hexacylated LPS could induce caspase-11 activation. However, this was not observed for tetracylated LPS [49]. The main effects of caspase-11 are associated with non-canonical pyroptosis [50]. During non-canonical pyroptosis, caspase-11 directly cleaves GSDMD as non-canonical inflammasome [51].
2.2.2. Gasdermin Family
Gasdermin (GSDM) is a family of pore-forming effector proteins discovered in recent years, and the GSDM family consists of Gasdermin A (GSDMA), Gasdermin B (GSDMB), Gasdermin C (GSDMC), Gasdermin D (GSDMD), Gasdermin E (GSDME, also called DFNA5), and pejvakin (PJVK, also called DFNB59) [3,52,53][3][52][53]. GSDM family members are expressed differently in different tissues and cells, and except for PJVK, all Gasdermins have conserved double domain arrangement: C-terminal domain and N-terminal domain, and the N-terminal domain has pore-forming activity [3,48,54,55][3][48][54][55]. The N-terminal domain of GSDMD is lipophilic and can bind with phosphatidylinositol phosphate, phosphatidylinoseroic acid, and cardiolipin, which makes the cell membrane form pores with the size of 10–14 nm, resulting in cell pyroptosis via the release of cell contents [8,56,57,58][8][56][57][58].
While GSDM family members are expressed differently in different tissues and cells, GSDMD is widely expressed in the cytosol of various cells and tissues [59]. It is the most important protein in the whole GSDM family as the common substrate protein of mouse caspase-1/11 or human caspase-4/5 and -1 [60]. GSDMD has 487 amino acids with 53 kDa weight and consists of 30 kDa N-terminal domain and 22 kDa C-terminal domain. Normally, C-terminal of GSDMD is connected to the N-terminal domain by a long loop, leaving GSDMD in an inactive autosuppressive state [56[56][59][61][62],59,61,62], while full-length GSDMD can be cleaved into two separate domains by caspase-1/11 in pyroptosis [59]; thereinto, N-terminal directly executes forming of membrane pore to induce pyroptosis [8]. However, neither C-terminal domain nor the full length GSDMD causes pyroptosis [61,62][61][62]. Consequently, GSDMD is the only substrate of inflammatory caspases, and the cleavage of GSDMD is a reliable marker of pyroptosis mediated by inflammatory caspases and inflammasome activation [8,58,63,64][8][58][63][64]. Therefore, GSDMD has become an important target for the intervention of pyroptosis.
2.2.3. Canonical and Noncanonical Inflammasome
The canonical inflammasome is a kind of multiprotein complex that mediates innate immune response in mammals. Its most prominent function is to recruit and activate caspase-1, promote the maturation of IL-1β and IL-18, and then generate inflammatory response. The inflammasome consists of NOD-like receptors (NLRs), apoptosis-associated Speck-like protein containing a CARD (ASC), and caspase-1 [65,66,67,68][65][66][67][68]. There are four intracellular receptor proteins to assemble the inflammasome, including NOD-like receptor protein 1 (NLRP1), NOD-like receptor protein 3 (NLRP3), NOD-like receptor C4 (NLRC4), and absent in melanoma 2 (AIM2) inflammasome [69]. The receptor is composed of three homologous domains: the N-terminal pyrin domain (PYD), the central nucleotide-binding oligomerization domain, and the C-terminal leucine repeat (LRR) [70]. In the process of canonical inflammasome assembly, NLRP3 recruits ASC through interaction with homotypic PYD domain and induces ASC to aggregate into macromolecular spots [71]. Subsequently, the assembled ASC recruits pro-caspase-1 through homotypic card domain interaction to form NLRP3-ASC-caspase-1 protein complex [72] that is known as NLRP3 inflammasome consisted of “sensor”, “adaptor”, and “effector” (Figure 2), respectively [73].
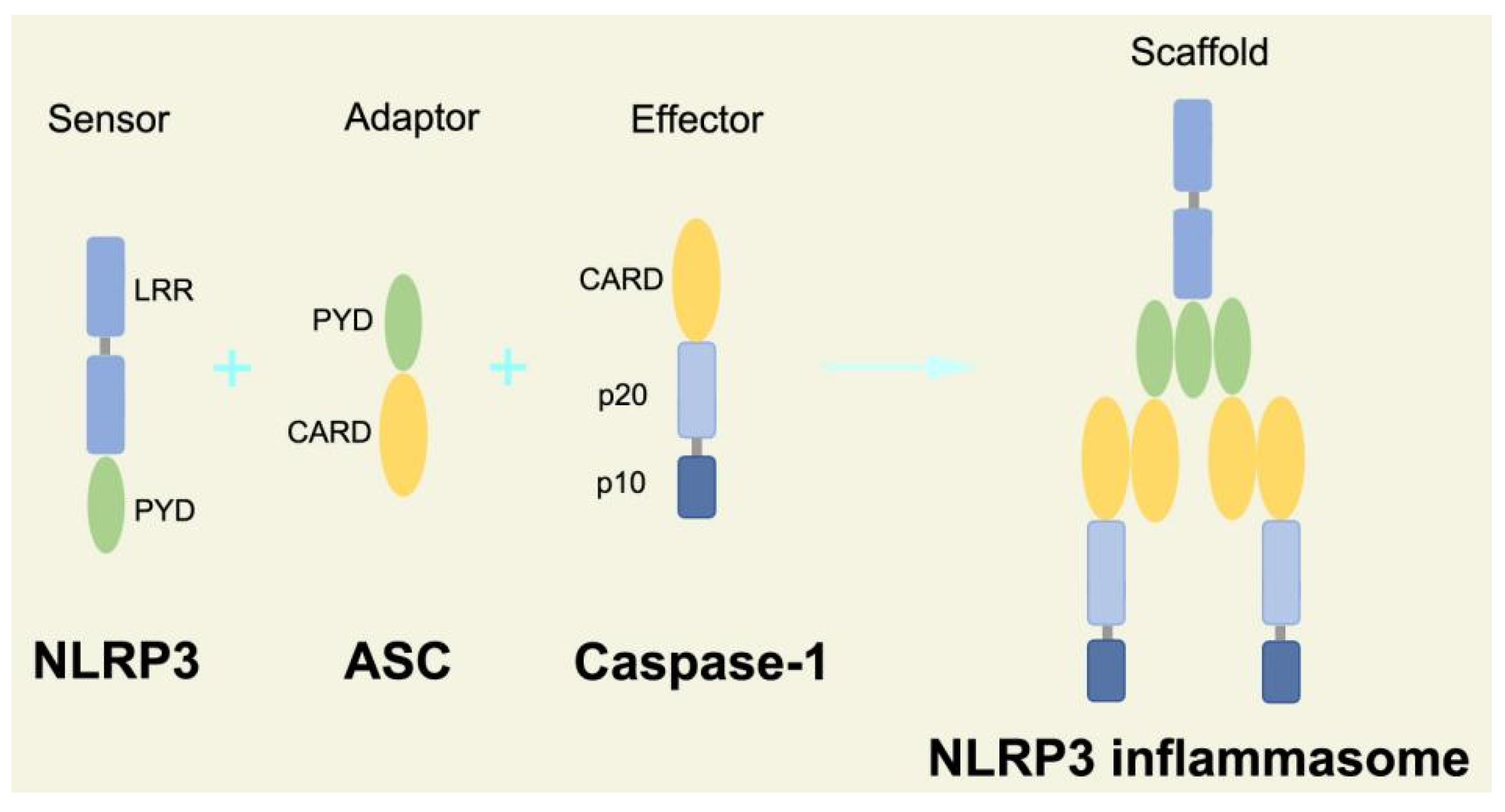
Figure 2. Structure and assembly of NLRP3 inflammasome. NLRP3 inflammasome consists of NLRP3, ASC, and caspase-1, which are known as sensor, adaptor, and effector, respectively.
In the canonical pathway of pyroptosis, when cytosolic pathogen recognition receptors (PRRs), NLRP1b, NLRP3, NLRC4, AIM2, or Pyrin are stimulated by the corresponding PAMPs and DAMPs, these proteins recruit ASC and pro-caspase-1 to assemble into inflammasomes. The NLRP3 inflammasome is described in detail, and its assembly and activation process is divided into two steps. Firstly, pathogen-associated molecular patterns (PAMPs) or damage-associated molecular patterns (DAMPs) are recognized by Toll-like receptor 4 (TLR4) and activate the nuclear factor kappa-B (NF-κB) pathway, leading to increased transcription of NLRP3, pro-caspase-1, pro-IL-1β, and pro-IL-18 [74]. Secondly, under the further stimulation of immune and inflammatory molecules, the NLRP3 protein is oligomerized and assembled with ASC and pro-caspase-1 to form the NLRP3 inflammasome [75]. Formation of NLRP3 inflammasome results in cleavage of pro-caspase-1 to form the active form of caspase-1 and promotes cleavage of pro-IL-1β and pro-IL-18 to form IL-1β and IL-18 mature bodies, leading to a cascade of immune or inflammatory responses [76].
In the noncanonical pathway of pyroptosis, the complex of LPS-pro-caspase-11 was recognized as a noncanonical inflammasome, and LPS directly activates caspase-11, which is independent to TLR4 signaling pathway [77]. When caspase-11 mediates noncanonical pyroptosis, it leads to cleavage of GSDMD, and GSDMD-N executes pyroptosis to induce the release of cell contents. However, noncanonical pyroptosis still requires the help of the NLRP3 inflammasome that activates caspase-1 to induce maturation of pro-IL-1β and pro-IL-18 [77].
2.3. Mechanism of Pyroptosis
2.3.1. Canonical Pyroptosis Pathway
Canonical pyroptosis is mediated by caspase-1, mainly in macrophages, and its key steps are the recruitment and activation of caspase-1 (Figure 3). Taking NLRP3 inflammasome as an example, when NLRP3 protein is stimulated by specific PAMPs and DAMPs, NLRP3 protein recruits ASC and pro-caspase-1 and assembles into NLRP3 inflammasome with the assistance of NIMA-related kinase 7 (NEK7) [78]. NLRP3 inflammasome assembly activates pro-caspase-1, and the activated caspase-1 can not only mediate the maturation and secretion of IL-1β and IL-18, but also directly cleave GSDMD to produce GSDMD-N [79]. Subsequently, GSDMD-N binds to phosphatidylinositol, phosphatidic acid, and phosphatidylserine on the inner surface of the membrane through membrane lipid interaction and forms oligomeric pores (GSDMS pore) with an inner diameter of 10~20 nm in the lipid bilayer. Then, LDH, IL-1β, and IL-18 are leaked out through the pores as well as other small cytosolic proteins and eventually cause pyroptosis [57]. Meanwhile, a large number of holes in the plasma membrane lead to the connection between the inner and outer membrane, forming a non-selective membrane channel, resulting in the efflux of K+ ions, which imbalance the ion concentration on both sides of the plasma membrane and cause a large amount of water to enter the cell, causing cell swelling and the eventual death of cells [8,80][8][80]. Under normal conditions, K+ efflux is generally considered to be both sufficient and necessary for NLRP3 inflammasome activation [81]. In addition, NLRC4, which is recognized by DAMPs or PAMPs, can directly activate caspase-1 to promote canonical pyroptosis [82].
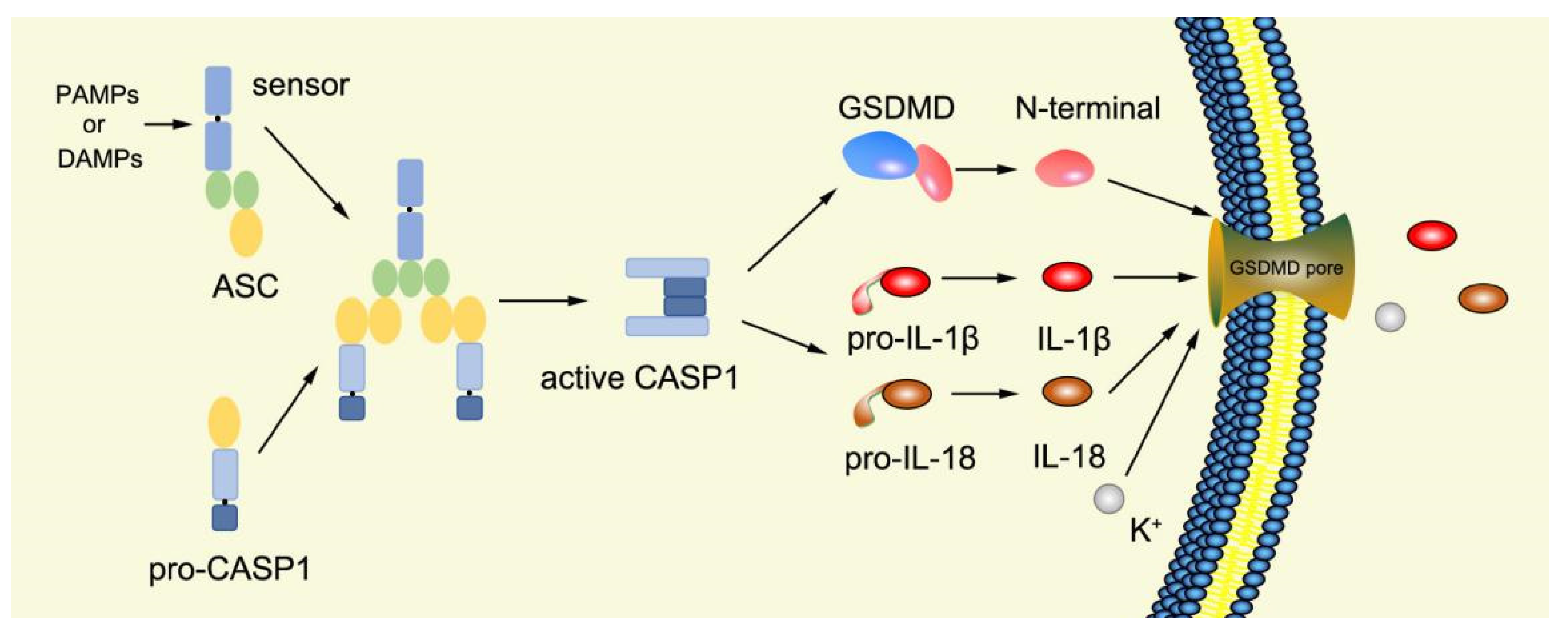
Figure 3. Mechanism of canonical pyroptosis pathway.
2.3.2. Noncanonical Pyroptosis Pathway
Numerous studies have found that caspase-1-independent pyroptosis pathway also exists in cells, which is named noncanonical pyroptosis and mediated by direct activation of caspase-4/5/11 under the action of LPS [83]. When Gram-negative bacteria infects mice, LPS is transferred by vesicles and enters the infected cells [84]. Mouse caspase-11 has CARD that can directly recognize the lipid A of LPS in the cytoplasm. After specific binding, caspase-11 oligomerized and activated, thereby mediating pyroptosis [48]. The functions of human caspase-4/5 and mouse caspase-11 are the same in mediating noncanonical pyroptosis [85]. Thus, caspase-4/5 also directly binds to LPS and promotes its own oligomerization and activation [86]. The activated caspase-4/5/11 can act on GSDMD and generate GSDMD-N fragments, thereby leading to cell membrane perforation and inducing pyroptosis [48,84,87,88,89][48][84][87][88][89]. In the pathway of canonical pyroptosis, caspase-1 cleaves IL-1β and IL-18 precursors to form active IL-1β and IL-18 and also cleaves GSDMD to produce N terminal; meanwhile, in the pathway of noncanonical pyroptosis, GSDMD cleavage is completed by the activated caspase-11 (Figure 4), but the cleavage of IL-1β and IL-18 precursor remains to depend on caspase-1 [43]. Thus, the noncanonical pyroptosis pathway depends on the activation of caspase-4/5/11 that lacks ASC participation, which is different from the canonical pathway. For human infection, activated caspase-4/5/11 can open Pannexin-1 channels to induce K+ efflux, which leads to NLRP3 inflammasome activation, promotes IL-1β and IL-18 maturation, and produces GSDMD-N to execute pyroptosis [48,79,83,90,91,92,93][48][79][83][90][91][92][93]. Meanwhile, ATP released from Pannexin-1 channels can activate P2X7R and promote K+ efflux, which in turn further promotes inflammasome assembly and triggers pyroptosis [78,94][78][94]. In addition, GSDMD-N can indirectly activate caspase-1 through the NLRP3-ASC-csapase-1 pathway, thereby promoting the maturation of IL-1β and IL-18 [95]. Therefore, new studies increasingly discover that caspase-4/5/11 is both a receptor and an effector molecule in the pathway of noncanonical pyroptosis [48].
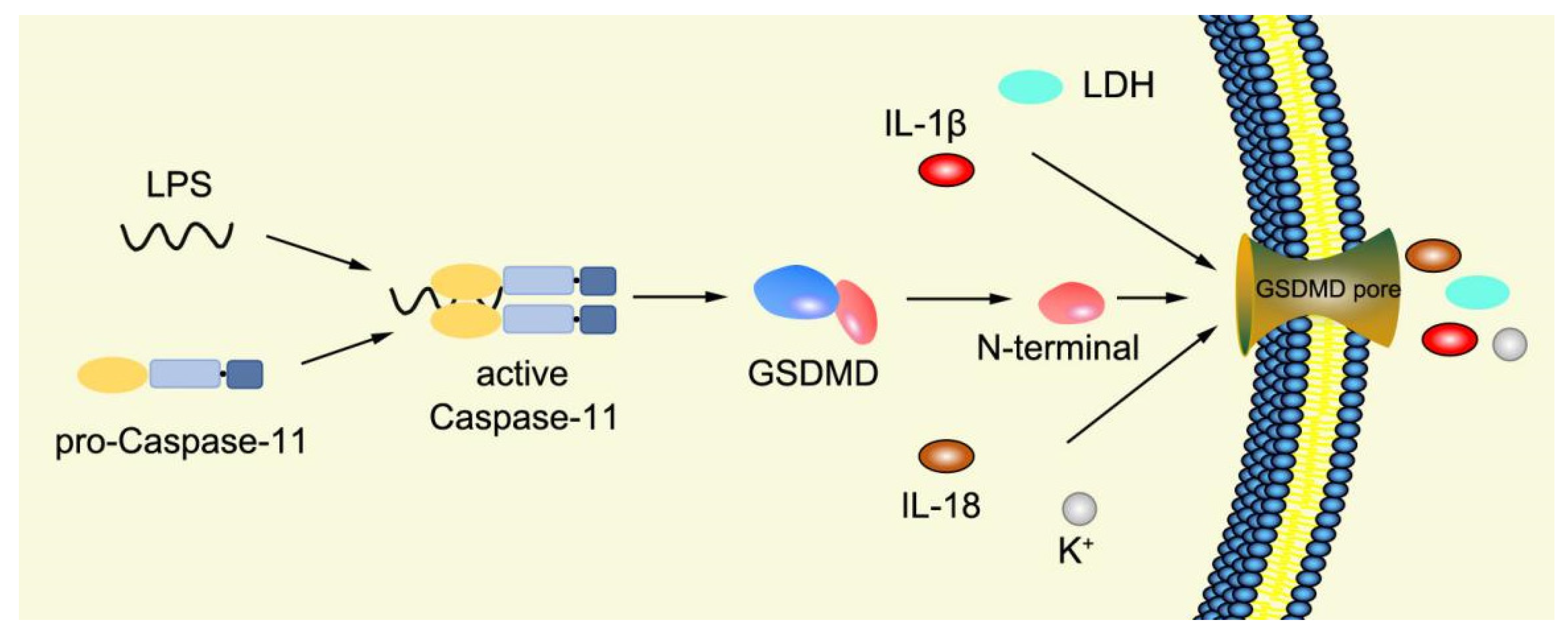
Figure 4. Mechanism of noncanonical pyroptosis pathway.
References
- Zychlinsky, A.; Prevost, M.C.; Sansonetti, P.J. Shigella flexneri induces apoptosis in infected macrophages. Nature 1992, 358, 167–169.
- Cookson, B.T.; Brennan, M.A. Pro-inflammatory programmed cell death. Trends Microbiol. 2001, 9, 113–114.
- Broz, P.; Pelegrin, P.; Shao, F. The gasdermins, a protein family executing cell death and inflammation. Nat. Rev. Immunol. 2020, 20, 143–157.
- Wu, C.; Lu, W.; Zhang, Y.; Zhang, G.; Shi, X.; Hisada, Y.; Grover, S.P.; Zhang, X.; Li, L.; Xiang, B.; et al. Inflammasome Activation Triggers Blood Clotting and Host Death through Pyroptosis. Immunity 2019, 50, 1401–1411.e4.
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016, 16, 407–420.
- Rathinam, V.A.K.; Zhao, Y.; Shao, F. Innate immunity to intracellular LPS. Nat. Immunol. 2019, 20, 527–533.
- Burdette, B.E.; Esparza, A.N.; Zhu, H.; Wang, S. Gasdermin D in pyroptosis. Acta Pharm. Sin. B 2021, 11, 2768–2782.
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665.
- Jorgensen, I.; Rayamajhi, M.; Miao, E.A. Programmed cell death as a defence against infection. Nat. Rev. Immunol. 2017, 17, 151–164.
- Vanden Berghe, T.; Hassannia, B.; Vandenabeele, P. An outline of necrosome triggers. Cell. Mol. Life Sci. 2016, 73, 2137–2152.
- Ge, X.; Li, W.; Huang, S.; Yin, Z.; Xu, X.; Chen, F.; Kong, X.; Wang, H.; Zhang, J.; Lei, P. The pathological role of NLRs and AIM2 inflammasome-mediated pyroptosis in damaged blood-brain barrier after traumatic brain injury. Brain. Res. 2018, 1697, 10–20.
- Kumar, S. Caspase function in programmed cell death. Cell Death Differ. 2007, 14, 32–43.
- Eckhart, L.; Ballaun, C.; Hermann, M.; VandeBerg, J.L.; Sipos, W.; Uthman, A.; Fischer, H.; Tschachler, E. Identification of novel mammalian caspases reveals an important role of gene loss in shaping the human caspase repertoire. Mol. Biol. Evol. 2008, 25, 831–841.
- Yin, Y.; Pastrana, J.L.; Li, X.; Huang, X.; Mallilankaraman, K.; Choi, E.T.; Madesh, M.; Wang, H.; Yang, X.F. Inflammasomes: Sensors of metabolic stresses for vascular inflammation. Front. Biosci. (Landmark Ed.) 2013, 18, 638–649.
- Crowley, L.C.; Waterhouse, N.J. Detecting Cleaved Caspase-3 in Apoptotic Cells by Flow Cytometry. Cold Spring Harb. Protoc. 2016, 2016, pdb-prot087312.
- Park, H.H. Structural features of caspase-activating complexes. Int. J. Mol. Sci. 2012, 13, 4807–4818.
- Zhaolin, Z.; Guohua, L.; Shiyuan, W.; Zuo, W. Role of pyroptosis in cardiovascular disease. Cell Prolif. 2019, 52, e12563.
- Black, R.A.; Kronheim, S.R.; Sleath, P.R. Activation of interleukin-1 beta by a co-induced protease. FEBS Lett. 1989, 247, 386–390.
- Kostura, M.J.; Tocci, M.J.; Limjuco, G.; Chin, J.; Cameron, P.; Hillman, A.G.; Chartrain, N.A.; Schmidt, J.A. Identification of a monocyte specific pre-interleukin 1 beta convertase activity. Proc. Natl. Acad. Sci. USA 1989, 86, 5227–5231.
- Cerretti, D.P.; Kozlosky, C.J.; Mosley, B.; Nelson, N.; Van Ness, K.; Greenstreet, T.A.; March, C.J.; Kronheim, S.R.; Druck, T.; Cannizzaro, L.A.; et al. Molecular cloning of the interleukin-1 beta converting enzyme. Science 1992, 256, 97–100.
- Thornberry, N.A.; Bull, H.G.; Calaycay, J.R.; Chapman, K.T.; Howard, A.D.; Kostura, M.J.; Miller, D.K.; Molineaux, S.M.; Weidner, J.R.; Aunins, J.; et al. A novel heterodimeric cysteine protease is required for interleukin-1 beta processing in monocytes. Nature 1992, 356, 768–774.
- Ghayur, T.; Banerjee, S.; Hugunin, M.; Butler, D.; Herzog, L.; Carter, A.; Quintal, L.; Sekut, L.; Talanian, R.; Paskind, M.; et al. Caspase-1 processes IFN-gamma-inducing factor and regulates LPS-induced IFN-gamma production. Nature 1997, 386, 619–623.
- Gu, Y.; Kuida, K.; Tsutsui, H.; Ku, G.; Hsiao, K.; Fleming, M.A.; Hayashi, N.; Higashino, K.; Okamura, H.; Nakanishi, K.; et al. Activation of interferon-gamma inducing factor mediated by interleukin-1beta converting enzyme. Science 1997, 275, 206–209.
- Fuentes-Prior, P.; Salvesen, G.S. The protein structures that shape caspase activity, specificity, activation and inhibition. Biochem. J. 2004, 384, 201–232.
- Roschitzki-Voser, H.; Schroeder, T.; Lenherr, E.D.; Frolich, F.; Schweizer, A.; Donepudi, M.; Ganesan, R.; Mittl, P.R.; Baici, A.; Grutter, M.G. Human caspases in vitro: Expression, purification and kinetic characterization. Protein Expr. Purif. 2012, 84, 236–246.
- Denault, J.B.; Salvesen, G.S. Human caspase-7 activity and regulation by its N-terminal peptide. J. Biol. Chem. 2003, 278, 34042–34050.
- Vaidya, S.; Velazquez-Delgado, E.M.; Abbruzzese, G.; Hardy, J.A. Substrate-induced conformational changes occur in all cleaved forms of caspase-6. J. Mol. Biol. 2011, 406, 75–91.
- Shi, Y. Mechanisms of caspase activation and inhibition during apoptosis. Mol. Cell 2002, 9, 459–470.
- Shi, Y. Caspase activation: Revisiting the induced proximity model. Cell 2004, 117, 855–858.
- Kersse, K.; Lamkanfi, M.; Bertrand, M.J.M.; Vanden Berghe, T.; Vandenabeele, P. Interaction patches of procaspase-1 caspase recruitment domains (CARDs) are differently involved in procaspase-1 activation and receptor-interacting protein 2 (RIP2)-dependent nuclear factor kappaB signaling. J. Biol. Chem. 2011, 286, 35874–35882.
- Boucher, D.; Monteleone, M.; Coll, R.C.; Chen, K.W.; Ross, C.M.; Teo, J.L.; Gomez, G.A.; Holley, C.L.; Bierschenk, D.; Stacey, K.J.; et al. Caspase-1 self-cleavage is an intrinsic mechanism to terminate inflammasome activity. J. Exp. Med. 2018, 215, 827–840.
- Ross, C.; Chan, A.H.; Von Pein, J.; Boucher, D.; Schroder, K. Dimerization and auto-processing induce caspase-11 protease activation within the non-canonical inflammasome. Life Sci. Alliance 2018, 1, e201800237.
- Feeney, B.; Clark, A.C. Reassembly of active caspase-3 is facilitated by the propeptide. J. Biol. Chem. 2005, 280, 39772–39785.
- Butt, A.J.; Harvey, N.L.; Parasivam, G.; Kumar, S. Dimerization and autoprocessing of the Nedd2 (caspase-2) precursor requires both the prodomain and the carboxyl-terminal regions. J. Biol. Chem. 1998, 273, 6763–6768.
- Van Criekinge, W.; Beyaert, R.; Van de Craen, M.; Vandenabeele, P.; Schotte, P.; De Valck, D.; Fiers, W. Functional characterization of the prodomain of interleukin-1beta-converting enzyme. J. Biol. Chem. 1996, 271, 27245–27248.
- Stennicke, H.R.; Deveraux, Q.L.; Humke, E.W.; Reed, J.C.; Dixit, V.M.; Salvesen, G.S. Caspase-9 can be activated without proteolytic processing. J. Biol. Chem. 1999, 274, 8359–8362.
- Ponder, K.G.; Boise, L.H. The prodomain of caspase-3 regulates its own removal and caspase activation. Cell Death Discov. 2019, 5, 56.
- Yang, X.; Chang, H.Y.; Baltimore, D. Autoproteolytic activation of pro-caspases by oligomerization. Mol. Cell 1998, 1, 319–325.
- Elliott, J.M.; Rouge, L.; Wiesmann, C.; Scheer, J.M. Crystal structure of procaspase-1 zymogen domain reveals insight into inflammatory caspase autoactivation. J. Biol. Chem. 2009, 284, 6546–6553.
- Wilson, K.P.; Black, J.A.; Thomson, J.A.; Kim, E.E.; Griffith, J.P.; Navia, M.A.; Murcko, M.A.; Chambers, S.P.; Aldape, R.A.; Raybuck, S.A.; et al. Structure and mechanism of interleukin-1 beta converting enzyme. Nature 1994, 370, 270–275.
- Datta, D.; McClendon, C.L.; Jacobson, M.P.; Wells, J.A. Substrate and inhibitor-induced dimerization and cooperativity in caspase-1 but not caspase-3. J. Biol. Chem. 2013, 288, 9971–9981.
- Martinon, F.; Mayor, A.; Tschopp, J. The inflammasomes: Guardians of the body. Annu. Rev. Immunol. 2009, 27, 229–265.
- Riedl, S.J.; Shi, Y. Molecular mechanisms of caspase regulation during apoptosis. Nat. Rev. Mol. Cell Biol. 2004, 5, 897–907.
- Renatus, M.; Stennicke, H.R.; Scott, F.L.; Liddington, R.C.; Salvesen, G.S. Dimer formation drives the activation of the cell death protease caspase 9. Proc. Natl. Acad. Sci. USA 2001, 98, 14250–14255.
- Salvesen, G.S.; Dixit, V.M. Caspase activation: The induced-proximity model. Proc. Natl. Acad. Sci. USA 1999, 96, 10964–10967.
- Boatright, K.M.; Salvesen, G.S. Mechanisms of caspase activation. Curr. Opin. Cell Biol. 2003, 15, 725–731.
- Kumar, S. Mechanisms mediating caspase activation in cell death. Cell Death Differ. 1999, 6, 1060–1066.
- Shi, J.; Zhao, Y.; Wang, Y.; Gao, W.; Ding, J.; Li, P.; Hu, L.; Shao, F. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 2014, 514, 187–192.
- Hagar, J.A.; Powell, D.A.; Aachoui, Y.; Ernst, R.K.; Miao, E.A. Cytoplasmic LPS activates caspase-11: Implications in TLR4-independent endotoxic shock. Science 2013, 341, 1250–1253.
- Agnew, A.; Nulty, C.; Creagh, E.M. Regulation, Activation and Function of Caspase-11 during Health and Disease. Int. J. Mol. Sci. 2021, 22, 1506.
- Broz, P.; von Moltke, J.; Jones, J.W.; Vance, R.E.; Monack, D.M. Differential requirement for Caspase-1 autoproteolysis in pathogen-induced cell death and cytokine processing. Cell Host Microbe 2010, 8, 471–483.
- Liu, X.; Xia, S.; Zhang, Z.; Wu, H.; Lieberman, J. Channelling inflammation: Gasdermins in physiology and disease. Nat. Rev. Drug Discov. 2021, 20, 384–405.
- Feng, S.; Fox, D.; Man, S.M. Mechanisms of Gasdermin Family Members in Inflammasome Signaling and Cell Death. J. Mol. Biol. 2018, 430 (Pt B), 3068–3080.
- Man, S.M.; Kanneganti, T.D. Gasdermin D: The long-awaited executioner of pyroptosis. Cell Res. 2015, 25, 1183–1184.
- England, H.; Summersgill, H.R.; Edye, M.E.; Rothwell, N.J.; Brough, D. Release of interleukin-1alpha or interleukin-1beta depends on mechanism of cell death. J. Biol. Chem. 2014, 289, 15942–15950.
- Liu, Z.; Wang, C.; Yang, J.; Zhou, B.; Yang, R.; Ramachandran, R.; Abbott, D.W.; Xiao, T.S. Crystal Structures of the Full-Length Murine and Human Gasdermin D Reveal Mechanisms of Autoinhibition, Lipid Binding, and Oligomerization. Immunity 2019, 51, 43–49.
- Sborgi, L.; Ruhl, S.; Mulvihill, E.; Pipercevic, J.; Heilig, R.; Stahlberg, H.; Farady, C.J.; Muller, D.J.; Broz, P.; Hiller, S. GSDMD membrane pore formation constitutes the mechanism of pyroptotic cell death. EMBO J. 2016, 35, 1766–1778.
- Liu, X.; Zhang, Z.; Ruan, J.; Pan, Y.; Magupalli, V.G.; Wu, H.; Lieberman, J. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 2016, 535, 153–158.
- Ding, J.; Wang, K.; Liu, W.; She, Y.; Sun, Q.; Shi, J.; Sun, H.; Wang, D.C.; Shao, F. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 2016, 535, 111–116.
- Ding, J.; Shao, F. Growing a Gasdermin Pore in Membranes of Pyroptotic Cells. EMBO J. 2018, 37, e100067.
- Liu, Z.; Wang, C.; Rathkey, J.K.; Yang, J.; Dubyak, G.R.; Abbott, D.W.; Xiao, T.S. Structures of the Gasdermin D C-Terminal Domains Reveal Mechanisms of Autoinhibition. Structure 2018, 26, 778–784.
- Xia, S. Biological Mechanisms and Therapeutic Relevance of the Gasdermin Family. Mol. Aspects Med. 2020, 76, 100890.
- Mulvihill, E.; Sborgi, L.; Mari, S.A.; Pfreundschuh, M.; Hiller, S.; Muller, D.J. Mechanism of Membrane Pore Formation by Human Gasdermin-D. EMBO J. 2018, 37, e98321.
- Zhao, Y.; Shi, J.; Shao, F. Inflammatory Caspases: Activation and Cleavage of Gasdermin-D in Vitro and During Pyroptosis. Methods Mol. Biol. 2018, 1714, 131–148.
- von Moltke, J.; Ayres, J.S.; Kofoed, E.M.; Chavarria-Smith, J.; Vance, R.E. Recognition of bacteria by inflammasomes. Annu. Rev. Immunol. 2013, 31, 73–106.
- Latz, E. The inflammasomes: Mechanisms of activation and function. Curr. Opin. Immunol. 2010, 22, 28–33.
- Lamkanfi, M.; Dixit, V.M. Inflammasomes and their roles in health and disease. Annu. Rev. Cell Dev. Biol. 2012, 28, 137–161.
- Skeldon, A.; Saleh, M. The Inflammasomes: Molecular Effectors of Host Resistance against Bacterial, Viral, Parasitic, and Fungal Infections. Front. Microbiol. 2011, 2, 15.
- Sun, C.; Dong, J.; Kuang, Z.; Yin, M.; Liu, X.; Deng, H. Research progresses on NLRP3 inflammasomes-induced anti-tumor immunity. Acta Pharm. Sin. 2022, 9, 2612–2621. (In Chinese)
- Ting, J.P.; Lovering, R.C.; Alnemri, E.S.; Bertin, J.; Boss, J.M.; Davis, B.K.; Flavell, R.A.; Girardin, S.E.; Godzik, A.; Harton, J.A.; et al. The NLR gene family: A standard nomenclature. Immunity 2008, 28, 285–287.
- Lu, A.; Magupalli, V.G.; Ruan, J.; Yin, Q.; Atianand, M.K.; Vos, M.R.; Schroder, G.F.; Fitzgerald, K.A.; Wu, H.; Egelman, E.H. Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell 2014, 156, 1193–1206.
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 2002, 10, 417–426.
- Agostini, L.; Martinon, F.; Burns, K.; McDermott, M.F.; Hawkins, P.N.; Tschopp, J. NALP3 forms an IL-1beta-processing inflammasome with increased activity in Muckle-Wells autoinflammatory disorder. Immunity 2004, 20, 319–325.
- Bauernfeind, F.G.; Horvath, G.; Stutz, A.; Alnemri, E.S.; MacDonald, K.; Speert, D.; Fernandes-Alnemri, T.; Wu, J.; Monks, B.G.; Fitzgerald, K.A.; et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J. Immunol. 2009, 183, 787–791.
- Biasizzo, M.; Kopitar-Jerala, N. Interplay between Nlrp3 Inflammasome and Autophagy. Front. Immunol. 2020, 11, 591803.
- Garlanda, C.; Dinarello, C.A.; Mantovani, A. The interleukin-1 family: Back to the future. Immunity 2013, 39, 1003–1018.
- Yang, D.; He, Y.; Munoz-Planillo, R.; Liu, Q.; Nunez, G. Caspase-11 Requires the Pannexin-1 Channel and the Purinergic P2X7 Pore to Mediate Pyroptosis and Endotoxic Shock. Immunity 2015, 43, 923–932.
- He, Y.; Zeng, M.Y.; Yang, D.; Motro, B.; Nunez, G. NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature 2016, 530, 354–357.
- He, W.T.; Wan, H.; Hu, L.; Chen, P.; Wang, X.; Huang, Z.; Yang, Z.H.; Zhong, C.Q.; Han, J. Gasdermin D is an executor of pyroptosis and required for interleukin-1beta secretion. Cell Res. 2015, 25, 1285–1298.
- Chen, X.; He, W.T.; Hu, L.; Li, J.; Fang, Y.; Wang, X.; Xu, X.; Wang, Z.; Huang, K.; Han, J. Pyroptosis is driven by non-selective gasdermin-D pore and its morphology is different from MLKL channel-mediated necroptosis. Cell Res. 2016, 26, 1007–1020.
- Christgen, S.; Place, D.E.; Kanneganti, T.D. Toward targeting inflammasomes: Insights into their regulation and activation. Cell Res. 2020, 30, 315–327.
- Case, C.L.; Roy, C.R. Asc modulates the function of NLRC4 in response to infection of macrophages by Legionella pneumophila. mBio 2011, 2, e00117-11.
- Kayagaki, N.; Warming, S.; Lamkanfi, M.; Vande Walle, L.; Louie, S.; Dong, J.; Newton, K.; Qu, Y.; Liu, J.; Heldens, S.; et al. Non-canonical inflammasome activation targets caspase-11. Nature 2011, 479, 117–121.
- Wang, M.; Jiang, S.; Zhang, Y.; Li, P.; Wang, K. The Multifaceted Roles of Pyroptotic Cell Death Pathways in Cancer. Cancers 2019, 11, 1313.
- Shi, J.; Gao, W.; Shao, F. Pyroptosis: Gasdermin-Mediated Programmed Necrotic Cell Death. Trends Biochem. Sci. 2017, 42, 245–254.
- He, Y.; Hara, H.; Nunez, G. Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends Biochem. Sci. 2016, 41, 1012–1021.
- Chen, Q.; Shi, P.; Wang, Y.; Zou, D.; Wu, X.; Wang, D.; Hu, Q.; Zou, Y.; Huang, Z.; Ren, J.; et al. GSDMB promotes non-canonical pyroptosis by enhancing caspase-4 activity. J. Mol. Cell Biol. 2019, 11, 496–508.
- Hsu, J.L.; Chou, J.W.; Chen, T.F.; Hsu, J.T.; Su, F.Y.; Lan, J.L.; Wu, P.C.; Hu, C.M.; Lee, E.Y.; Lee, W.H. Glutathione peroxidase 8 negatively regulates caspase-4/11 to protect against colitis. EMBO Mol. Med. 2020, 12, e9386.
- Yang, J.; Zhao, Y.; Shao, F. Non-canonical activation of inflammatory caspases by cytosolic LPS in innate immunity. Curr. Opin. Immunol. 2015, 32, 78–83.
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O’Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.; Haley, B.; Roose-Girma, M.; Phung, Q.T.; et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 2015, 526, 666–671.
- Sarhan, J.; Liu, B.C.; Muendlein, H.I.; Li, P.; Nilson, R.; Tang, A.Y.; Rongvaux, A.; Bunnell, S.C.; Shao, F.; Green, D.R.; et al. Caspase-8 induces cleavage of gasdermin D to elicit pyroptosis during Yersinia infection. Proc. Natl. Acad. Sci. USA 2018, 115, E10888–E10897.
- Ruhl, S.; Broz, P. Caspase-11 activates a canonical NLRP3 inflammasome by promoting K(+) efflux. Eur. J. Immunol. 2015, 45, 2927–2936.
- Huang, Y.; Xu, W.; Zhou, R. NLRP3 inflammasome activation and cell death. Cell. Mol. Immunol. 2021, 18, 2114–2127.
- Xu, Z.; Chen, Z.M.; Wu, X.; Zhang, L.; Cao, Y.; Zhou, P. Distinct Molecular Mechanisms Underlying Potassium Efflux for NLRP3 Inflammasome Activation. Front. Immunol. 2020, 11, 609441.
- Martinon, F.; Tschopp, J. Inflammatory caspases: Linking an intracellular innate immune system to autoinflammatory diseases. Cell 2004, 117, 561–574.
More