Medin, a small 50-amino acid peptide, is an internal cleaved product from the second discoidin domain of milk fat globule epidermal growth factor VIII (MFG-E8) protein. Medin has been reported as the most common amylogenic protein in the upper part of the arterial system, including aortic, temporal, and cerebral arterial walls in the elderly. Medin has a high affinity to elastic fibers and is closely associated with arterial degenerative inflammation, elastic fiber fragmentation, calcification, and amyloidosis. In vitro, treating with the medin peptide promotes the inflammatory phenotypic shift of both endothelial cells and vascular smooth muscle cells. In vitro, ex vivo, and in vivo studies demonstrate that medin enhances the abundance of reactive oxygen species and reactive nitrogen species produced by both endothelial cells and vascular smooth muscle cells and promotes vascular endothelial dysfunction and arterial stiffening. Immunostaining and immunoblotting analyses of human samples indicate that the levels of medin are increased in the pathogenesis of aortic aneurysm/dissection, temporal arteritis, and cerebrovascular dementia. Thus, medin peptide could be targeted as a biomarker diagnostic tool or as a potential molecular approach to curbing the arterial degenerative inflammatory remodeling that accompanies aging and disease.
- medin
- inflammation
- disease
1. Medin Induces Vascular Cell Inflammation
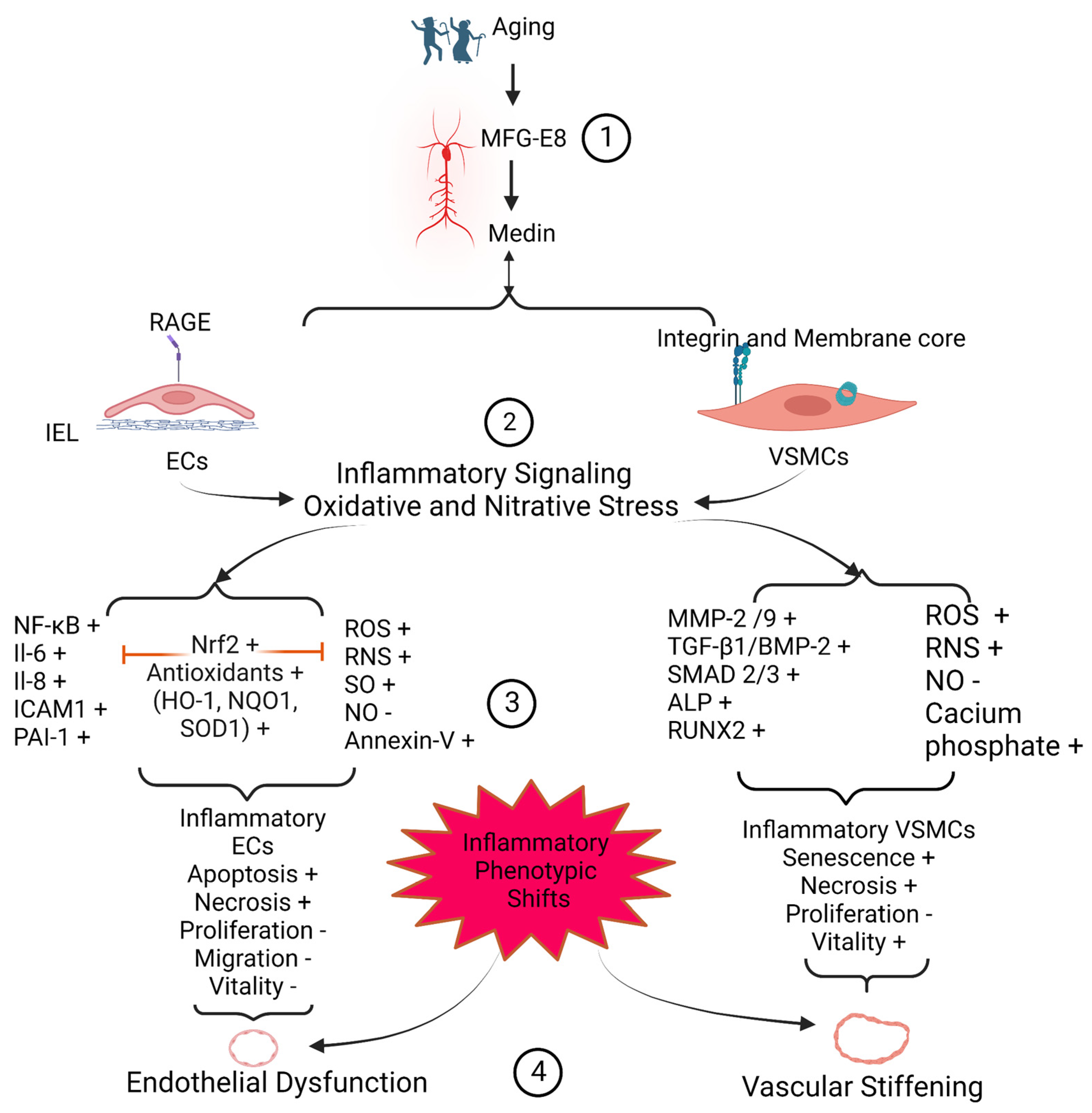
1.1. Endothelial Cells Inflammation and Viability
1.2. Vascular SMCmooth Muscle Cells Inflammation and Viability
2. Medin Deposition Impairs Vascular Function
The remodeled or modified vascular ECM and inflammatory vascular cells contribute to the dysfunction of the vascular wall. The internal elastin laminae have a high affinity to medin peptide, forming amyloids [14,17,22][20][21][22]. The medin-associated internal elastic lamina greatly impacts on the microenvironment of the overlying endothelial layer, promoting the structural and functional deterioration, likely contributing to endothelial dysfunction in larger and smaller arteries (Figure 1 and Figure 2). Amyloid internal elastic fibers are potentially susceptible to fracture and the loss of elasticity; and this stiffened physical barrier is harmful to the survival of ECs and susceptible to the medial invasion of VSMCs, likely leading to VSMC infiltration of the subendothelial space; and this stiffened physical barrier also changes its chemical property, such as with the increased accumulation of oxidized low-density lipoproteins in fractured internal elastic ends [3][7]. All these alterations may contribute to the stiffening of the vascular wall (Figure 1 and Figure 2).
2.1. Arterial Endothelial Disorder and Dysfunction
Aging increases endothelial disorder and dysfunction in the arterial wall. Endothelial-dependent vasodilation declines with advancing age in both men and women [3][7]. Aging impairs the endothelial physical layer via endothelial apoptosis and senescence and increases the endothelial cellular gap due to alterations of endothelial cellular junctions, facilitating increases in inflammation, thrombosis, and permeability [3][7]. In large arteries, endothelial dysfunction also promotes intimal medial thickening, arterial stiffening, and increases in blood pressure [3][7]. In the small arteries such as in the brain, aging damages the cerebrovascular endothelial layer, promoting inflammation, thrombosis, permeability, micro-bleeding/thrombosis/embolism, and decreases the blood perfusion, leading to an insufficient supply of nutrients and oxygen and producing reactive oxidized/nitrative species (ROS/RNS), contributing to neurovascular unit structural and functional damage [1,2,4,13,27,28][2][4][13][15][23][24]. Medin treatment significantly damaged the endothelial-dependent dilation of human leptomeningeal arterioles [9,28][2][25]. Importantly, genetic deficiency of the medin precursor protein, MFG-E8, eliminates not only vascular medin aggregates but also prevents the age-associated decline of cerebrovascular endothelial dependent dysfunction [1][13]. Accumulating evidence indicates that medin is involved in the detrimental effects on arterial endothelial-dependent dilation and exerts an important role in the development of endothelial dysfunction with aging through increased EC apoptosis, oxidative stress, inflammation and or the decrease in NO bioavailability as well as the EC survival ability [2,22,28,33,46][2][4][6][22][26].2.2. Arterial Microstructure Disorder and Stiffening
The pulse wave velocity (PWV), a clinical gold standard measure of arterial stiffness, increases with age in both men and women [3][7]. Arterial stiffening could be attributed to the increases in both vascular cells and matrix stiffness with advancing age. Atomic force microscopy (AFM) observations indicate that the stiffness of both elastic laminae and the inter-space of elastic laminae are increased with aging [55,56,57][27][28][29]. Furthermore, in vitro studies demonstrate that both ECs and VSMCs isolated from old animals become stiffer than those from young animals [58,59,60,61,62][30][31][32][33][34]. An oscillatory nanoindentation measurement can localize and determine the local vascular wall mechanical properties via the measurement of shear storage modulus, G’ and shear loss modulus, G˝ [22]. This examination using this nanoindentation technique shows that significantly lower G’, an index of reduction of elasticity of the tissue, has been detected in the elastin-contractile units, enriched with MFG-E8, medin, oligomer medin and medin amyloid fibrils [22]. In other words, the higher MFG-E8, oligomer medin, medin amyloid fibrils in the elastin-contractile units in the arterial wall convey higher levels of local vascular microstructure stiffness [22]. Thus, an increase in medin amyloid greatly increases vascular wall stiffness, contributing to increasing blood pressure, aneurysm/dissection, cerebrovascular dysfunction, and cognition decline in the elderly.3. Medin Deposition Is Involved in the Pathogenesis of Vascular Diseases
Medin has been reported to be elevated in aortic aneurysm/dissection, temporal arteritis, and vascular dementia [2,4,17,24][4][16][21][23]. Medin-associated vascular cell apoptosis and necrosis; medin-associated elastin degeneration, calcification, and amyloidosis result in a weakness of the vascular wall. Importantly, medin-associated endothelial dysfunction and arterial wall stiffening results in an imbalance of mechanical force on the local arterial wall. This medin-associated structural and function remodeling contributes to the pathogenesis of arterial aneurysm and dissection (Figure 52). The internal elastin laminae bound to the medin amyloid, trigger endothelial immune reactivation, leading to the dysfunction of the arterioles in the brain. Importantly, endothelial dysfunction causes an insufficient supply of blood to brain tissue by cerebrovascular tissue, leading to the brain ischemia and abnormal immune activation, eventually contributing to the pathogenesis of vascular-related dementia (Figure 52). In addition, medin and the associated fragment elastin peptide are engulfed by macrophages, causing giant cell vasculitis (Figure 2).3.1. Arterial Aneurysms and Dissections
Arterial aneurysm and dissections are usually a complication of advanced stage hypertension, atherosclerosis, and diabetes [63][35]. Arterial aneurysm is an excessive focal enlargement/expansion of the artery caused by the weakening of the arterial wall. Arterial dissection is an abnormal, and usually abrupt, formation of a tear (intimal) along the inside wall of an artery [63][35]. The pathologic foundations in the arterial wall aneurysm/dissection are closely associated with the degradation of the intima and media, which includes EC, VSMCs, collagen, elastin, and elastin-contractile units [63][35]. Aneurysm and dissection areas become inflamed, stiffened, and fragile, which are closely associated to a disorder of cellular proteostasis and the quality of protein [63][35]. Interestingly, MFG-E8, its fragments and medin-derived amyloids, have been detected in patients afflicted by either thoracic aortic aneurysms or type A aortic dissection [22,24,29][16][22][36]. The abundance of MFG-E8, which produces medin, is elevated in the aortic media of older-aged subjects where amyloidosis is enhanced [1,24,40][13][16][19]. Previous findings suggest that MFG-E8 seems to be decreased in the aortic aneurysm or dissection while medin and medin amyloid are increased [11,22,24,63][16][22][35][37]. Remarkably, the number of senescent VSMCs is markedly increased in the aortic aneurysm/dissection which secrete extracellular vesicles, containing MFG-E8 and medin, promoting medin amyloid formation [37,64][12][38]. It has been reported that medin aggregates into amyloid, leading to potentially fatal conditions of thoracic aortic aneurysm and dissection [22,24,29][16][22][36]. Notably, medin is increased along with the biomarkers of oxidative stress 8-hydroxy-2’-deoxyguanosine and 4-hydroxy-2-nonenal in the aortic media of middle-aged or older-aged donors [7][1]. Oxidative damage induces the disruption of VSMCs, resulting in the decrease of α-actin, a highly expressed protein in contractile VSMCs, and matrix remodeling, causing the vulnerability of the aging arterial wall [30,32][3][39]. In addition, medin was the conspicuous trigger for necrosis in VSMCs and for the release of activated MMP-2, thus further promoting the degradation of the aortic matrix, facilitating the development of aneurysm/dissection [22,24][16][22]. The senescence and necrosis of VSMCs and the breakdown of elastic fiber networks is the pathologic foundation of the development of aortic aneurysm or dissection. These findings suggest that targeting MFG-E8 or its fragment medin is a novel molecular approach to diagnose, prevent, or treat aortic aneurysm/dissection.3.2. Temporal Arteritis
Temporal arteritis, also known as giant cell arteritis, is an inflammatory disease of the blood vessels near the temple [65,66][40][41]. The prevalence of temporal arteritis ranges from approximately 0.5 to 27 cases per 100,000 people, aged 50 years or older [66,67][41][42]. In the clinic, temporal arteritis can cause damage to eyesight, including sudden blindness in one or both eyes, or damage other arterioles, leading to aneurysm/dissection, stroke, or a transient ischemic attack [65][40]. Medin amyloid deposits commonly occur along the internal elastic lamina of the temporal artery in the elderly [16,17][21][43]. In temporal artery biopsies from 22 patients with clinical and histological signs of giant cell arteritis, medin amyloid deposits were found in 14 (64%) biopsies [17][21]. Furthermore, it is interesting that, under a microscope, the medin amyloid appeared topographically closely related to the elastin-derived fragments [12,17][14][21]. Notably, fragmented elastic material was often immunolabelled for medin and found to be engulfed by giant cells known as macrophages [12,17][14][21]. Furthermore, in situ hybridization showed that MFG-E8 was expressed locally by VSMCs of the temporal artery [12,17][14][21]. These findings suggest that MFG-E8 or its fragment medin play a pathologic role in the inflammatory process in giant cell arteritis. Thus, targeting medin or MFG-E8 is a novel potential molecular approach to the diagnosis, prevention and treatment of temporal arteritis.3.3. Vascular-Related Cognitive Impairment and Dementia
The endothelial dysfunction of the cerebrovascular system plays an important role in age-associated vascular contributions to cognitive impairment and dementia (VCID [2,9,68,69,70][4][25][44][45][46]). VCID is a decline in thinking capacity caused by conditions that block or reduce cerebrovascular blood flow or perfusion to various regions of the brain, depriving them of oxygen and nutrients [9,68,69,70][25][44][45][46]. Insufficient blood flow, ischemia, can damage and kill cells in the tissue, especially in the vulnerable brain in the elderly, and eventually alter their thinking skills [68,70,71][44][46][47]. Thinking defects can start as mild ischemia that gradually worsen because of multiple minor strokes, microbleeds or micro-embolisms that affect smaller blood vessels, promoting the widespread damage of a vascular-neuro unit and causing VCID [72,73][48][49]. The prevalence of VCID exponentially increases with advancing age in modern society. Notably, VCID is ranked as the second most frequent cause of dementia, exceeded only by Alzheimer’s disease (AD). Vascular and cellular senescence is closely associated with cognition decline with aging [74,75][50][51]. VSMC senescence accelerates medin aggregation through small extracellular vesicle secretion and extracellular matrix reorganization [37,42][12][52]. A growing body of evidence documents that amyloid deposition is closely associated with cerebrovascular medin abundance in humans; and the medin amyloid load is higher in vascular dementia patients than in cognitively normal patients [2,4,9,28][2][4][23][25]. Medin amyloid is a key element of the neurovascular unit in the brain, causing microvascular endothelial dysfunction through oxidative and nitrative stress, and promotes inflammatory signaling in EC via RAGE, showing EC immune activation and neuroinflammation [2,9][4][25]. In addition, medin induces EC immune activation via the release of interleukin-6/-8 (IL-6/8), which subsequently modulates astrocyte inflammatory activation [2,28][2][4]. These immune reactions and inflammation promote the development of degenerative aneurysm and microstructure weakening [1][13]. This localized structure and functional inflammatory remodeling contributes to local ischemia in the brain and eventually causes cognitive decline [1][13]. Importantly, the medin-induced endothelial dysfunction and oxidative stress are markedly reversed by the antioxidant polyethylene glycol superoxide dismutase or by the RAGE inhibitor, FPS-ZM1 [2,28,33][2][4][6]. A recent study shows that aggregates of medin have been found in the brain vasculature of wild-type mice in an age-dependent manner and are closely associated with age-related cognitive decline [1][13]. Strikingly, genetic deficiency of the medin precursor protein, MFG-E8, eliminates not only vascular aggregates but also prevents age-associated decline of cerebrovascular function in mice [1][13]. Remarkably, cerebrovascular medin also is a strong predictor of AD [4][23], and a recent study demonstrates that medin peptide forms amyloids in cerebrovascular walls and promotes vascular Aβ deposits by fibrilizing them with Aβ; in contrast, lowering medin protects against cerebral amyloid angiopathy and cognition impairment in mice by a cross-seeding mechanism [27][24]. Thus, medin could be a biomarker diagnostic tool for AD.References
- Miura, Y.; Tsumoto, H.; Iwamoto, M.; Yamaguchi, Y.; Ko, P.; Soejima, Y.; Yoshida, S.; Toda, T.; Arai, T.; Hamamatsu, A.; et al. Age-associated proteomic alterations in human aortic media. Geriatr. Gerontol. Int. 2019, 19, 1054–1062.
- Migrino, R.Q.; Davies, H.A.; Truran, S.; Karamanova, N.; Franco, D.A.; Beach, T.G.; Serrano, G.E.; Truong, D.; Nikkhah, M.; Madine, J. Amyloidogenic medin induces endothelial dysfunction and vascular inflammation through the receptor for advanced glycation endproducts. Cardiovasc. Res. 2017, 113, 1389–1402.
- Davies, H.A.; Phelan, M.M.; Wilkinson, M.C.; Migrino, R.Q.; Truran, S.; Franco, D.A.; Liu, L.N.; Longmore, C.J.; Madine, J. Oxidative Stress Alters the Morphology and Toxicity of Aortic Medial Amyloid. Biophys. J. 2015, 109, 2363–2370.
- Karamanova, N.; Truran, S.; Serrano, G.E.; Beach, T.G.; Madine, J.; Weissig, V.; Davies, H.A.; Veldhuizen, J.; Nikkhah, M.; Hansen, M.; et al. Endothelial Immune Activation by Medin: Potential Role in Cerebrovascular Disease and Reversal by Monosialoganglioside-Containing Nanoliposomes. J. Am. Heart Assoc. 2020, 9, e014810.
- Madine, J.; Middleton, D.A. Comparison of aggregation enhancement and inhibition as strategies for reducing the cytotoxicity of the aortic amyloid polypeptide medin. Eur. Biophys. J. 2010, 39, 1281–1288.
- Karamanova, N.; Truran, S.; Weissig, V.; Madine, J.; Davies, H.; Guzman-Villanueva, D.; Migrino, R. Amyloidogenic Medin Impairs Endothelial Cell Autophagy and Viability that is Reversed by Monosialoganglioside Nanoliposomes. FASEB J. 2018, 32, 846.816.
- Wang, M.; Jiang, L.; Monticone, R.E.; Lakatta, E.G. Proinflammation: The key to arterial aging. Trends Endocrinol. Metab. 2014, 25, 72–79.
- Wang, M.; Monticon, R.; McGraw, K. Proinflammation, profibrosis, and arterial aging. Aging Med. 2020, 3, 159–168.
- Li, B.Y.; Li, X.L.; Cai, Q.; Gao, H.Q.; Cheng, M.; Zhang, J.H.; Wang, J.F.; Yu, F.; Zhou, R.H. Induction of lactadherin mediates the apoptosis of endothelial cells in response to advanced glycation end products and protective effects of grape seed procyanidin B2 and resveratrol. Apoptosis 2011, 16, 732–745.
- Li, B.Y.; Li, X.L.; Gao, H.Q.; Zhang, J.H.; Cai, Q.; Cheng, M.; Lu, M. Grape seed procyanidin B2 inhibits advanced glycation end product-induced endothelial cell apoptosis through regulating GSK3beta phosphorylation. Cell Biol. Int. 2011, 35, 663–669.
- Nerelius, C.; Gustafsson, M.; Nordling, K.; Larsson, A.; Johansson, J. Anti-amyloid activity of the C-terminal domain of proSP-C against amyloid beta-peptide and medin. Biochemistry 2009, 48, 3778–3786.
- Whitehead, M.; Yusoff, S.; Ahmad, S.; Schmidt, L.; Mayr, M.; Madine, J.; Middleton, D.; Shanahan, C.M. Vascular smooth muscle cell senescence accelerates medin aggregation via small extracellular vesicle secretion and extracellular matrix reorganization. Aging Cell 2022, e13746.
- Degenhardt, K.; Wagner, J.; Skodras, A.; Candlish, M.; Koppelmann, A.J.; Wild, K.; Maxwell, R.; Rotermund, C.; von Zweydorf, F.; Gloeckner, C.J.; et al. Medin aggregation causes cerebrovascular dysfunction in aging wild-type mice. Proc. Natl. Acad. Sci. USA 2020, 117, 23925–23931.
- Haggqvist, B.; Naslund, J.; Sletten, K.; Westermark, G.T.; Mucchiano, G.; Tjernberg, L.O.; Nordstedt, C.; Engstrom, U.; Westermark, P. Medin: An integral fragment of aortic smooth muscle cell-produced lactadherin forms the most common human amyloid. Proc. Natl. Acad. Sci. USA 1999, 96, 8669–8674.
- Larsson, A.; Malmstrom, S.; Westermark, P. Signs of cross-seeding: Aortic medin amyloid as a trigger for protein AA deposition. Amyloid 2011, 18, 229–234.
- Peng, S.; Larsson, A.; Wassberg, E.; Gerwins, P.; Thelin, S.; Fu, X.; Westermark, P. Role of aggregated medin in the pathogenesis of thoracic aortic aneurysm and dissection. Lab. Invest. 2007, 87, 1195–1205.
- Davies, H.A.; Madine, J.; Middleton, D.A. Comparisons with amyloid-beta reveal an aspartate residue that stabilizes fibrils of the aortic amyloid peptide medin. J. Biol. Chem. 2015, 290, 7791–7803.
- Ni, Y.-Q.; Li, S.; Lin, X.; Wang, Y.-J.; He, J.-Y.; Song, W.-L.; Xiang, Q.-Y.; Zhao, Y.; Li, C.; Wang, Y.; et al. Exosomal MFGE8 from high glucose induced endothelial cells is involved in calcification/senescence of vascular smooth muscle cells. bioRxiv 2021.
- Younger, S.; Jang, H.; Davies, H.A.; Niemiec, M.J.; Garcia, J.G.N.; Nussinov, R.; Migrino, R.Q.; Madine, J.; Arce, F.T. Medin Oligomer Membrane Pore Formation: A Potential Mechanism of Vascular Dysfunction. Biophys. J. 2020, 118, 2769–2782.
- Larsson, A.; Peng, S.; Persson, H.; Rosenbloom, J.; Abrams, W.R.; Wassberg, E.; Thelin, S.; Sletten, K.; Gerwins, P.; Westermark, P. Lactadherin binds to elastin--a starting point for medin amyloid formation? Amyloid 2006, 13, 78–85.
- Peng, S.; Westermark, G.T.; Naslund, J.; Haggqvist, B.; Glennert, J.; Westermark, P. Medin and medin-amyloid in ageing inflamed and non-inflamed temporal arteries. J. Pathol. 2002, 196, 91–96.
- Davies, H.A.; Caamano-Gutierrez, E.; Chim, Y.H.; Field, M.; Nawaytou, O.; Ressel, L.; Akhtar, R.; Madine, J. Idiopathic degenerative thoracic aneurysms are associated with increased aortic medial amyloid. Amyloid 2019, 26, 148–155.
- Migrino, R.Q.; Karamanova, N.; Truran, S.; Serrano, G.E.; Davies, H.A.; Madine, J.; Beach, T.G. Cerebrovascular medin is associated with Alzheimer’s disease and vascular dementia. Alzheimer’s Dement. 2020, 12, e12072.
- Wagner, J.; Degenhardt, K.; Veit, M.; Louros, N.; Konstantoulea, K.; Skodras, A.; Wild, K.; Liu, P.; Obermüller, U.; Bansal, V.; et al. Medin co-aggregates with vascular amyloid-β in Alzheimer’s disease. Nature 2022, 612, 123–131.
- Migrino, R.Q.; Truran, S.; Karamanova, N.; Serrano, G.E.; Madrigal, C.; Davies, H.A.; Madine, J.; Reaven, P.; Beach, T.G. Human cerebral collateral arteriole function in subjects with normal cognition, mild cognitive impairment, and dementia. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H284–H290.
- Olofsson, A.; Borowik, T.; Grobner, G.; Sauer-Eriksson, A.E. Negatively charged phospholipid membranes induce amyloid formation of medin via an alpha-helical intermediate. J. Mol. Biol. 2007, 374, 186–194.
- Ushiki, T. Collagen fibers, reticular fibers and elastic fibers. A comprehensive understanding from a morphological viewpoint. Arch. Histol. Cytol. 2002, 65, 109–126.
- Raspanti, M.; Protasoni, M.; Manelli, A.; Guizzardi, S.; Mantovani, V.; Sala, A. The extracellular matrix of the human aortic wall: Ultrastructural observations by FEG-SEM and by tapping-mode AFM. Micron 2006, 37, 81–86.
- Berquand, A.; Wahart, A.; Henry, A.; Gorisse, L.; Maurice, P.; Blaise, S.; Romier-Crouzet, B.; Pietrement, C.; Bennasroune, A.; Sartelet, H.; et al. Revealing the elasticity of an individual aortic fiber during ageing at nanoscale by in situ atomic force microscopy. Nanoscale 2021, 13, 1124–1133.
- Kuznetsova, T.G.; Starodubtseva, M.N.; Yegorenkov, N.I.; Chizhik, S.A.; Zhdanov, R.I. Atomic force microscopy probing of cell elasticity. Micron 2007, 38, 824–833.
- Le Master, E.; Fancher, I.S.; Lee, J.; Levitan, I. Comparative analysis of endothelial cell and sub-endothelial cell elastic moduli in young and aged mice: Role of CD36. J. Biomech. 2018, 76, 263–268.
- Jannatbabaei, A.; Tafazzoli-Shadpour, M.; Seyedjafari, E. Effects of substrate mechanics on angiogenic capacity and nitric oxide release in human endothelial cells. Ann. N. Y. Acad. Sci. 2020, 1470, 31–43.
- Kolodziejczyk, A.M.; Kucinska, M.; Jakubowska, A.; Sokolowska, P.; Rosowski, M.; Tkacz-Szczesna, B.; Komorowski, P.; Makowski, K.; Walkowiak, B. Endothelial cell aging detection by means of atomic force spectroscopy. J. Mol. Recognit. 2020, 33, e2853.
- Zhu, W.; Kim, B.C.; Wang, M.; Huang, J.; Isak, A.; Bexiga, N.M.; Monticone, R.; Ha, T.; Lakatta, E.G.; An, S.S. TGFβ1 reinforces arterial aging in the vascular smooth muscle cell through a long-range regulation of the cytoskeletal stiffness. Sci. Rep. 2018, 8, 2668.
- Patel, P.D.; Arora, R.R. Pathophysiology, diagnosis, and management of aortic dissection. Ther. Adv. Cardiovasc. Dis. 2008, 2, 439–468.
- Hinterseher, I.; Erdman, R.; Elmore, J.R.; Stahl, E.; Pahl, M.C.; Derr, K.; Golden, A.; Lillvis, J.H.; Cindric, M.C.; Jackson, K.; et al. Novel pathways in the pathobiology of human abdominal aortic aneurysms. Pathobiology 2013, 80, 1–10.
- Westermark, G.T.; Westermark, P. Localized amyloids important in diseases outside the brain--lessons from the islets of Langerhans and the thoracic aorta. FEBS J. 2011, 278, 3918–3929.
- Chen, H.Z.; Wang, F.; Gao, P.; Pei, J.F.; Liu, Y.; Xu, T.T.; Tang, X.; Fu, W.Y.; Lu, J.; Yan, Y.F.; et al. Age-Associated Sirtuin 1 Reduction in Vascular Smooth Muscle Links Vascular Senescence and Inflammation to Abdominal Aortic Aneurysm. Circ. Res. 2016, 119, 1076–1088.
- Hu, R.; Wang, M.Q.; Ni, S.H.; Wang, M.; Liu, L.Y.; You, H.Y.; Wu, X.H.; Wang, Y.J.; Lu, L.; Wei, L.B. Salidroside ameliorates endothelial inflammation and oxidative stress by regulating the AMPK/NF-kappaB/NLRP3 signaling pathway in AGEs-induced HUVECs. Eur. J. Pharmacol. 2020, 867, 172797.
- Muniz Castro, H.M.; Bhattacharjee, M.B.; Chaudhry, I.A.; Chuang, A.Z.; Mankiewicz, K.A.; Adesina, O.O. Diagnosis of giant cell arteritis using clinical, laboratory, and histopathological findings in patients undergoing temporal artery biopsy. Clin. Neurol. Neurosurg. 2022, 221, 107377.
- Sharma, A.; Mohammad, A.J.; Turesson, C. Incidence and prevalence of giant cell arteritis and polymyalgia rheumatica: A systematic literature review. Semin. Arthritis Rheum. 2020, 50, 1040–1048.
- Weyand, C.M.; Goronzy, J.J. Medium- and large-vessel vasculitis. N. Engl. J. Med. 2003, 349, 160–169.
- Peng, S.; Glennert, J.; Westermark, P. Medin-amyloid: A recently characterized age-associated arterial amyloid form affects mainly arteries in the upper part of the body. Amyloid 2005, 12, 96–102.
- Wang, F.; Cao, Y.; Ma, L.; Pei, H.; Rausch, W.D.; Li, H. Dysfunction of Cerebrovascular Endothelial Cells: Prelude to Vascular Dementia. Front. Aging Neurosci. 2018, 10, 376.
- Ott, A.; Breteler, M.M.; van Harskamp, F.; Claus, J.J.; van der Cammen, T.J.; Grobbee, D.E.; Hofman, A. Prevalence of Alzheimer’s disease and vascular dementia: Association with education. The Rotterdam study. BMJ 1995, 310, 970–973.
- Caruso, P.; Signori, R.; Moretti, R. Small vessel disease to subcortical dementia: A dynamic model, which interfaces aging, cholinergic dysregulation and the neurovascular unit. Vasc. Health Risk. Manag. 2019, 15, 259–281.
- Guo, S.; Deng, W.; Xing, C.; Zhou, Y.; Ning, M.; Lo, E.H. Effects of aging, hypertension and diabetes on the mouse brain and heart vasculomes. Neurobiol. Dis. 2019, 126, 117–123.
- Knopman, D.S.; Parisi, J.E.; Boeve, B.F.; Cha, R.H.; Apaydin, H.; Salviati, A.; Edland, S.D.; Rocca, W.A. Vascular dementia in a population-based autopsy study. Arch. Neurol. 2003, 60, 569–575.
- McAleese, K.E.; Alafuzoff, I.; Charidimou, A.; De Reuck, J.; Grinberg, L.T.; Hainsworth, A.H.; Hortobagyi, T.; Ince, P.; Jellinger, K.; Gao, J.; et al. Post-mortem assessment in vascular dementia: Advances and aspirations. BMC Med. 2016, 14, 129.
- Bryant, A.G.; Hu, M.; Carlyle, B.C.; Arnold, S.E.; Frosch, M.P.; Das, S.; Hyman, B.T.; Bennett, R.E. Cerebrovascular Senescence Is Associated With Tau Pathology in Alzheimer’s Disease. Front. Neurol. 2020, 11, 575953.
- Ungvari, Z.; Tarantini, S.; Sorond, F.; Merkely, B.; Csiszar, A. Mechanisms of Vascular Aging, A Geroscience Perspective: JACC Focus Seminar. J. Am. Coll. Cardiol. 2020, 75, 931–941.
- Whitehead, M.; Ahmad, S.; Shanahan, C. BS47 Role of vascular smooth muscle cell-derived exosomes in age-related vascular amyloidosis. Heart 2019, 105, A169–A170.