Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Conner Chen and Version 3 by Conner Chen.
Platelets are the second most abundant blood component after red blood cells and can participate in a variety of physiological and pathological functions. Beyond its traditional role in hemostasis and thrombosis, it also plays an indispensable role in inflammatory diseases.
- toll-like receptors
- inflammation
- thrombopoiesis
1. Introduction
Platelets are small (2–4 μm) and are the end products of membrane protrusions from MKs that extend into the sinusoidal vessels, where they are sheered off by blood flow [1]. Remarkably, megakaryocytes (MKs) migrate from the HSC (hematopoietic stem cell) osteoblastic niche to the vascular niche, which is spatially and temporally regulated by transcription factors, signaling and adhesion molecules, and cytokines and chemokines in the process of progressive differentiation [2]. Various aspects, such as differentiation maturity, determine the formation of platelets. Generally, megakaryocytes tailor their cytoplasm and membrane systems for platelet biogenesis. Before a megakaryocyte has the capacity to release platelets, it enlarges considerably and fills with high concentrations of ribosomes and a complete demarcation membrane system (DMS) [3][4][5]. In adults, platelet generation can be divided into two stages that entail the differentiation of HSCs into mature megakaryocytes (MKs; termed megakaryocytes) and the release of platelets from MKs (termed thrombopoiesis or platelet biogenesis) (Figure 1) [6][7]. Although human adults contain nearly one trillion platelets in circulation that are turned over every 8–10 days, it is worth noting that the reduction in platelet count and the dysfunction of platelets will lead to a variety of diseases. Thus, the relevant mechanisms driving platelet formation and platelet function are appreciated. At present, platelets have been recognized as integral players in the inflammatory response and an important mediator in the activation of innate immunity [8][9][10][11][12]. At the same time, studies have shown that platelets express a variety of receptors, some of which are involved in platelet activation, platelet–leucocyte reciprocal activation, immunopathology, and platelet-dependent antimicrobial activity, including the TLR family [13][14]. Toll-like receptors (TLRs) can regulate inflammation and activate the innate immune signaling pathways. Meanwhile, TLRs exist in hematopoietic stem cells and megakaryocytes, which can promote platelet generation and function (see below).
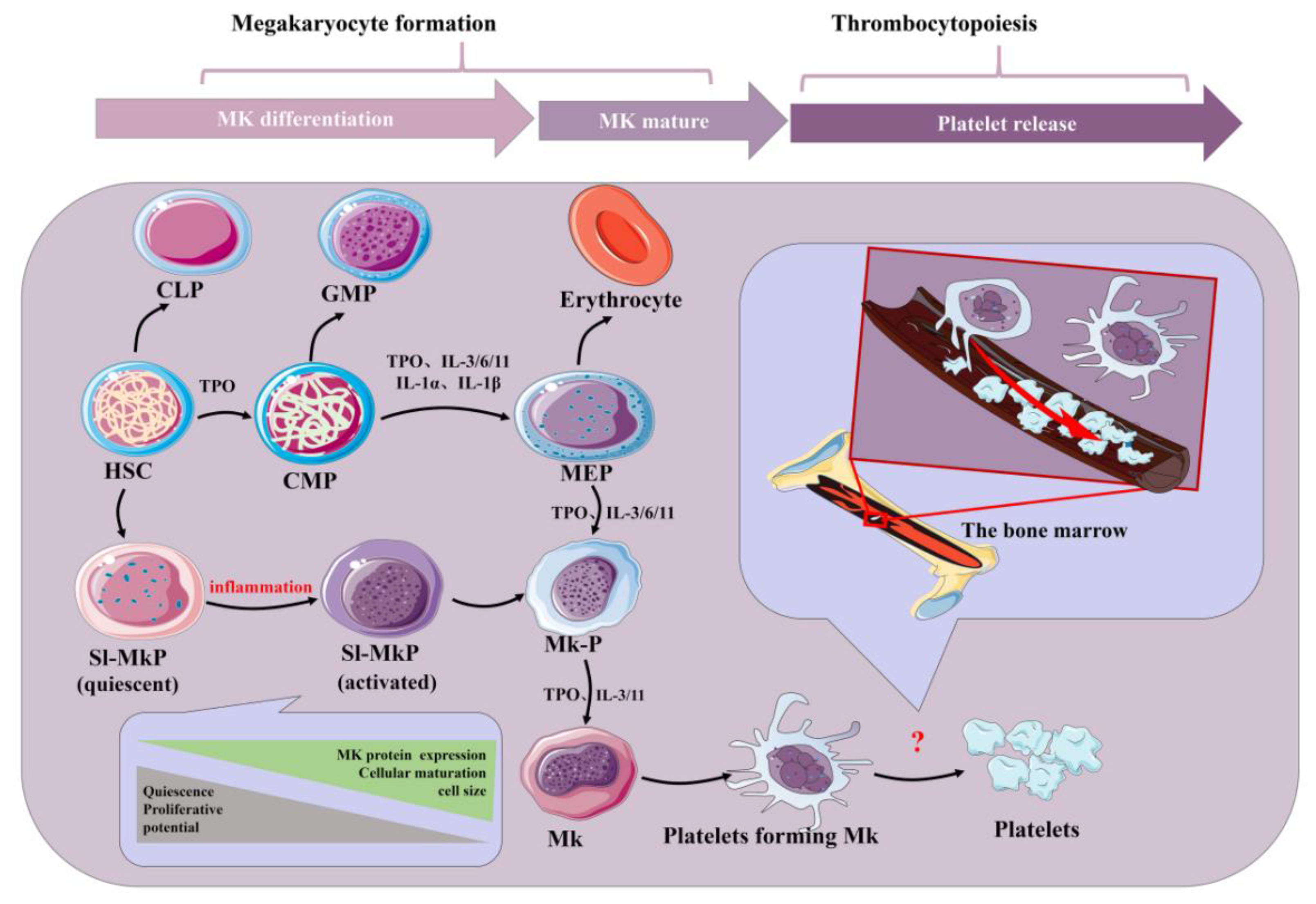
Figure 1. Schematic of platelet production. Inflammatory factors play an important role in the formation of platelets by bone marrow hematopoietic stem cells under physiological and inflammatory conditions. Acute inflammation can drive efficient cell cycle activation and maturation of SL-MKPs to rapidly replenish platelet pools. HSC: hematopoietic stem cell; CLP: common lymphoid progenitor; GMP: granulocyte-macrophage progenitor; CMP: common myeloid progenitor; MEP: megakaryocyte-erythroid progenitor; Mk-P: megakaryocyte progenitor; Sl-Mkp: stem-like megakaryocyte progenitors; TPO: thrombopoietin; MK: megakaryocyte; IL: interleukin.
TLRs are one of the most important receptors in the activation of innate immunity and have a certain regulatory effect on the regulation of inflammation, and there is an inseparable connection between the two [15]. TLR activation triggers signaling cascades with a common downstream signaling pathway to induce the production of pro-inflammatory cytokines and type I interferons: the former triggers the synthesis of the inflammatory mediators that cause fever, pain and other inflammation, and the latter mediates antiviral responses [16][17]. In addition, studies have found that TLR’s activation of the immune response is a temporary coordinated output that can transition from releasing pro-inflammatory attack mediators to initiating the inflammatory mediators required for decomposition and tissue repair [18]. However, the abnormal activation of TLR signals can lead to autoimmune, sepsis and chronic inflammatory diseases [19][20][21]. In conclusion, TLRs are an important therapeutic or regulatory target in the inflammatory response. On one hand, the high expression of TLR on CD34+ cells suggests that the expression of TLR on HSCs is an early sensory mechanism for HSCs to directly detect infection and immediately generate immune effector cells [22]. On the other hand, the expression of TLRs is also regulated during HSC differentiation, which has also been reported previously [23][24]. It has been proven that the inflammatory factors IL-3, IL-6, IL-9 and IL-11 indirectly affect TPO-induced MK production, while IL-3 and TPO synergistically promote MK differentiation [25][26][27]. Surprisingly, as early as 1987, an increase in MK progenitor cell formation was observed in mice treated with an inflammatory agent [28]. At the same time, TLR activation produces IL-6 and tumor necrosis factor-α (TNF-α). These two cytokines have a certain effect on platelet production. A series of studies have been conducted on its mechanism [29].
These results indicate that inflammatory factors can promote platelet production and that TLR is an important receptor for regulating inflammation. Meanwhile, platelets also express TLRs, and activating this receptor can regulate the function of platelets. In addition, TLR2 and TLR4 are the main receptors associated with platelet production/function and inflammation in the TLR family [30]. Moreover, some drugs acting on the TLR2 and TLR4 signaling pathways have been developed to treat diseases in order to improve the probability of cure and quality of life [31].
2. Inflammation and Platelets
Inflammation, as the second line of defense in innate immunity, is the body’s defense mechanism against pathogens, allergens/irritants, damaged cells, or any foreign invaders, manifested as redness, swelling, heat, pain, or dysfunction [32][33][34]. It is also an important part of innate immunity. At present, studies have shown that the body activates different pathways in response to the relevant stimuli and generally releases a large number of inflammatory factors, primarily γ-interferon, interleukin (IL), nitric oxide (NO) and TNF-α. It is beneficial to the body to remove harmful signals and initiate the response of the tissue healing process. Generally, it can be divided into two types, chronic and acute, depending on the characteristics of the response and stimulation. Among them, acute inflammation is primarily a form of regulation to restore homeostasis after damage to the body tissue, the symptoms of which are fever, redness, swelling and pain. The body response mainly involves mast cells releasing histamine and prostaglandins to dilate their blood vessels, and granulocytes migrate quickly to neutralize and remove harmful substances [35]. However, if acute inflammation fails to restore homeostasis, acute inflammation will gradually evolve into chronic inflammation and even cause more diseases. In chronic inflammation, macrophages and T lymphocytes mainly produce cytokines and enzymes that lead to tissue destruction, manifested as tissue fibrosis. As vascular first responders, platelets are key elements in the early phases of the inflammatory response (Figure 2). Platelets in the human body are not a uniform population, and platelet content varies with their size. Smaller platelets carry more inflammatory transcripts than larger platelets in a healthy population [36]. During the period of infection, antiviral protein in the blood platelet increases because the megakaryocyte cells start the reaction in the process of infection as a response to pathogens, directly activating platelets and releasing extracellular vesicles. In addition, platelets can directly sense pathogens and endogenous danger signals and promote the formation of antimicrobial and procoagulant neutrophil extracellular traps (NETs) through the expression of TLRs [37]. Remarkably, at the same time, platelets help leukocytes infiltrate tissues and exert their immune functions; they limit host tissue collateral damage by repairing vascular breaches caused by leukocyte trafficking and activation [37]. This suggests that platelets are an important mediator between innate immunity and inflammation [38]. In addition, the pathogenesis of inflammatory disease models has been demonstrated to rely on platelet-neutrophil complex formation [39]. First, they activate macrophages and monocytes and are critical in the recruitment and activation of neutrophils and they collaborate with neutrophils to initiate intravascular thrombosis, contributing to pathogen recognition, trapping, and disposal, thereby protecting the integrity of the host [39]. However, intravascular thrombosis caused by the interaction of platelets and white blood cells is called immune thrombosis, and this thrombotic reaction lasts too long in the body and may lead to disseminated intravascular coagulation (DIC), followed by multiple organ failure and even death. Second, platelets can migrate, aggregate, and adhere to bacteria in the body, contributing to the recruitment and stimulation of neutrophils and thus playing a direct antibacterial role [40][41]. This makes platelets attractive therapeutic targets for diseases ranging from chronic to acute inflammation or autoimmune diseases to bacterial, viral, and parasitic infections [42][43]. However, it is worth noting that, whilst platelets are the link between inflammation and host defense, the overreaction of platelets can also lead to disease. This is sufficient to show the multiplicity of platelets in inflammation.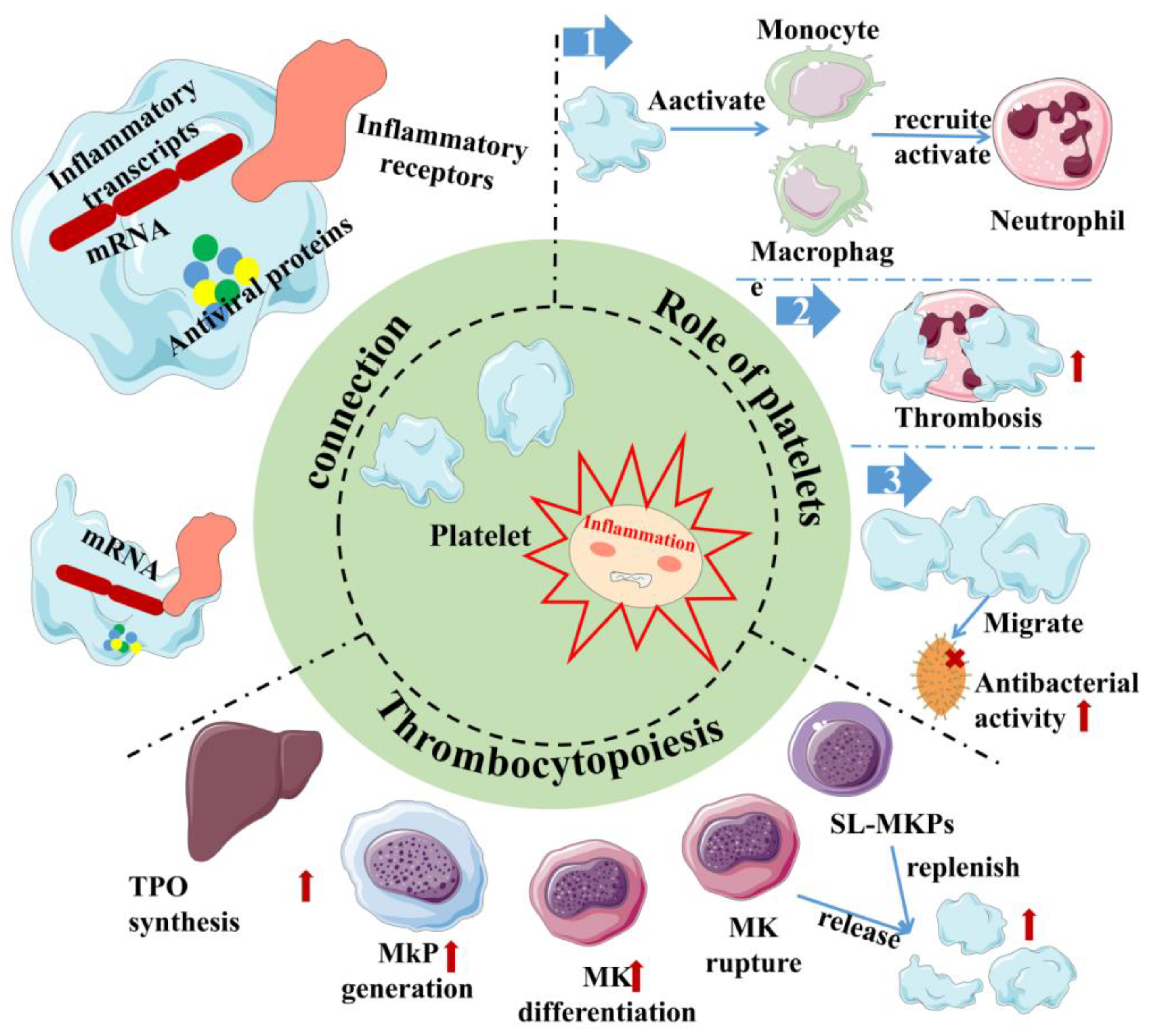
Figure 2. Inflammation and platelet interaction. Platelets are at the junction between inflammation and host defense. Platelets carry not only inflammatory transcripts but also antiviral proteins. Platelets activate/recruit a variety of cells for host defense in inflammation, meanwhile, and inflammation can promote platelet production through multiple channels. Red arrows indicate the enhanced physiological effects. Red “×” indicate the role of sterilization. Sl-Mkp: Stem-like megakaryocyte progenitors; TPO: Thrombopoietin; MK: megakaryocyte; MkP: Megakaryocyte progenitor.
References
- Thon, J.N.; Italiano, J.E. Platelets: Production, morphology and ultrastructure. In Antiplatelet Agents; Springer: Berlin/Heidelberg, Germany, 2012; pp. 3–22.
- Eto, K.; Kunishima, S. Linkage between the mechanisms of thrombocytopenia and thrombopoiesis. Blood 2016, 127, 1234–1241.
- Long, M.W.; Williams, N.; Ebbe, S. Immature megakaryocytes in the mouse: Physical characteristics, cell cycle status, and in vitro responsiveness to thrombopoietic stimulatory factor. Blood 1982, 59, 569–575.
- Yamada, E. The fine structure of the megakaryocyte in the mouse spleen. Cells Tissues Organs 1957, 29, 267–290.
- Nakao, K.; Angrist, A.A. Membrane surface specialization of blood platelet and megakaryocyte. Nature 1968, 217, 960–961.
- Noetzli, L.J.; French, S.L.; Machlus, K.R. New Insights Into the Differentiation of Megakaryocytes From Hematopoietic Progenitors. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1288–1300.
- Bennett, C.; Lawrence, M.; Guerrero, J.A.; Stritt, S.; Waller, A.K.; Yan, Y.; Mifsud, R.W.; Ballester-Beltrán, J.; Baig, A.; Mueller, A.; et al. CRLF3 plays a key role in the final stage of platelet genesis and is a potential therapeutic target for thrombocythemia. Blood 2022, 139, 2227–2239.
- Boilard, E.; Nigrovic, P.A.; Larabee, K.; Watts, G.F.; Coblyn, J.S.; Weinblatt, M.E.; Massarotti, E.M.; Remold-O’Donnell, E.; Farndale, R.W.; Ware, J.; et al. Platelets amplify inflammation in arthritis via collagen-dependent microparticle production. Science 2010, 327, 580–583.
- Huo, Y.; Schober, A.; Forlow, S.B.; Smith, D.F.; Hyman, M.C.; Jung, S.; Littman, D.R.; Weber, C.; Ley, K. Circulating activated platelets exacerbate atherosclerosis in mice deficient in apolipoprotein E. Nat. Med. 2003, 9, 61–67.
- McMorran, B.J.; Marshall, V.M.; de Graaf, C.; Drysdale, K.E.; Shabbar, M.; Smyth, G.K.; Corbin, J.E.; Alexander, W.S.; Foote, S.J. Platelets kill intraerythrocytic malarial parasites and mediate survival to infection. Science 2009, 323, 797–800.
- Semple, J.W.; Italiano, J.E., Jr.; Freedman, J. Platelets and the immune continuum. Nat. Rev. Immunol. 2011, 11, 264–274.
- Garraud, O.; Cognasse, F. Are Platelets Cells? And if Yes, are They Immune Cells? Front. Immunol. 2015, 6, 70.
- Nicolai, L.; Massberg, S. Platelets as key players in inflammation and infection. Curr. Opin. Hematol. 2020, 27, 34–40.
- Dib, P.R.B.; Quirino-Teixeira, A.C.; Merij, L.B.; Pinheiro, M.B.M.; Rozini, S.V.; Andrade, F.B.; Hottz, E.D. Innate immune receptors in platelets and platelet-leukocyte interactions. J. Leukoc. Biol. 2020, 108, 1157–1182.
- Yoshimura, A.; Ohishi, H.M.; Aki, D.; Hanada, T. Regulation of TLR signaling and inflammation by SOCS family proteins. J. Leukoc. Biol. 2004, 75, 422–427.
- Iwasaki, A.; Medzhitov, R. Toll-like receptor control of the adaptive immune responses. Nat. Immunol. 2004, 5, 987–995.
- Asami, J.; Shimizu, T. Structural and functional understanding of the toll-like receptors. Protein Sci. 2021, 30, 761–772.
- Mogensen, T.H. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin. Microbiol. Rev. 2009, 22, 240–273.
- Lai, Y.; Gallo, R.L. Toll-like receptors in skin infections and inflammatory diseases. Infect. Disord. Drug Targets 2008, 8, 144–155.
- Ospelt, C.; Gay, S. TLRs and chronic inflammation. Int. J. Biochem. Cell Biol. 2010, 42, 495–505.
- Joosten, L.A.; Abdollahi-Roodsaz, S.; Dinarello, C.A.; O’Neill, L.; Netea, M.G. Toll-like receptors and chronic inflammation in rheumatic diseases: New developments. Nat. Rev. Rheumatol. 2016, 12, 344–357.
- Baldridge, M.T.; King, K.Y.; Goodell, M.A. Inflammatory signals regulate hematopoietic stem cells. Trends Immunol. 2011, 32, 57–65.
- Dorner, M.; Brandt, S.; Tinguely, M.; Zucol, F.; Bourquin, J.P.; Zauner, L.; Berger, C.; Bernasconi, M.; Speck, R.F.; Nadal, D. Plasma cell toll-like receptor (TLR) expression differs from that of B cells, and plasma cell TLR triggering enhances immunoglobulin production. Immunology 2009, 128, 573–579.
- Takami, M.; Kim, N.; Rho, J.; Choi, Y. Stimulation by toll-like receptors inhibits osteoclast differentiation. J. Immunol. 2002, 169, 1516–1523.
- Jiang, H.; Yu, Z.; Ding, N.; Yang, M.; Zhang, L.; Fan, X.; Zhou, Y.; Zou, Q.; Hou, J.; Zheng, J.; et al. The role of AGK in thrombocytopoiesis and possible therapeutic strategies. Blood 2020, 136, 119–129.
- Kaushansky, K.; Broudy, V.C.; Lin, N.; Jorgensen, M.J.; McCarty, J.; Fox, N.; Zucker-Franklin, D.; Lofton-Day, C. Thrombopoietin, the Mp1 ligand, is essential for full megakaryocyte development. Proc. Natl. Acad. Sci. USA 1995, 92, 3234–3238.
- Panuganti, S.; Schlinker, A.C.; Lindholm, P.F.; Papoutsakis, E.T.; Miller, W.M. Three-stage ex vivo expansion of high-ploidy megakaryocytic cells: Toward large-scale platelet production. Tissue Eng. Part A 2013, 19, 998–1014.
- Clark, D.A.; Dessypris, E.N.; Koury, M.J. Induction of megakaryocytic colony-stimulating activity in mouse skin by inflammatory agents and tumor promoters. Proc. Soc. Exp. Biol. Med. 1987, 184, 245–249.
- Dan, K.; Gomi, S.; Inokuchi, K.; Ogata, K.; Yamada, T.; Ohki, I.; Hasegawa, S.; Nomura, T. Effects of interleukin-1 and tumor necrosis factor on megakaryocytopoiesis: Mechanism of reactive thrombocytosis. Acta Haematol. 1995, 93, 67–72.
- Semeraro, F.; Ammollo, C.T.; Morrissey, J.H.; Dale, G.L.; Friese, P.; Esmon, N.L.; Esmon, C.T. Extracellular histones promote thrombin generation through platelet-dependent mechanisms: Involvement of platelet TLR2 and TLR4. Blood 2011, 118, 1952–1961.
- Wang, Y.; Zhang, S.; Li, H.; Wang, H.; Zhang, T.; Hutchinson, M.R.; Yin, H.; Wang, X. Small-Molecule Modulators of Toll-like Receptors. Acc. Chem. Res. 2020, 53, 1046–1055.
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435.
- Vincenzo, B.; Asif, I.J.; Nikolaos, P.; Francesco, M. Adaptive immunity and inflammation. Int. J. Inflam. 2015, 2015, 575406.
- Newton, K.; Dixit, V.M. Signaling in innate immunity and inflammation. Cold Spring Harb. Perspect. Biol. 2012, 4, a006049.
- Maskrey, B.H.; Megson, I.L.; Whitfield, P.D.; Rossi, A.G. Mechanisms of resolution of inflammation: A focus on cardiovascular disease. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1001–1006.
- Clancy, L.; Beaulieu, L.M.; Tanriverdi, K.; Freedman, J.E. The role of RNA uptake in platelet heterogeneity. Thromb. Haemost. 2017, 117, 948–961.
- Wienkamp, A.K.; Erpenbeck, L.; Rossaint, J. Platelets in the NETworks interweaving inflammation and thrombosis. Front. Immunol. 2022, 13, 953129.
- Rayes, J.; Watson, S.P. Platelet GPVI repairs its own damage. Blood 2015, 126, 933–934.
- Massberg, S.; Grahl, L.; von Bruehl, M.L.; Manukyan, D.; Pfeiler, S.; Goosmann, C.; Brinkmann, V.; Lorenz, M.; Bidzhekov, K.; Khandagale, A.B.; et al. Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nat. Med. 2010, 16, 887–896.
- Rosowski, E.E.; Huttenlocher, A. Motile Collectors: Platelets Promote Innate Immunity. Immunity 2018, 48, 16–18.
- Bambach, S.K.; Lämmermann, T. Platelets, On Your Marks, Get Set, Migrate! Cell 2017, 171, 1256–1258.
- Sexton, T.R.; Zhang, G.; Macaulay, T.E.; Callahan, L.A.; Charnigo, R.; Vsevolozhskaya, O.A.; Li, Z.; Smyth, S. Ticagrelor Reduces Thromboinflammatory Markers in Patients with Pneumonia. JACC Basic Transl. Sci. 2018, 3, 435–449.
- Kor, D.J.; Carter, R.E.; Park, P.K.; Festic, E.; Banner-Goodspeed, V.M.; Hinds, R.; Talmor, D.; Gajic, O.; Ware, L.B.; Gong, M.N. Effect of Aspirin on Development of ARDS in At-Risk Patients Presenting to the Emergency Department: The LIPS-A Randomized Clinical Trial. JAMA 2016, 315, 2406–2414.
- Kaushansky, K.; Lok, S.; Holly, R.D.; Broudy, V.C.; Lin, N.; Bailey, M.C.; Forstrom, J.W.; Buddle, M.M.; Oort, P.J.; Hagen, F.S.; et al. Promotion of megakaryocyte progenitor expansion and differentiation by the c-Mpl ligand thrombopoietin. Nature 1994, 369, 568–571.
- Trinh, B.Q.; Barengo, N.; Kim, S.B.; Lee, J.S.; Zweidler-McKay, P.A.; Naora, H. The homeobox gene DLX4 regulates erythro-megakaryocytic differentiation by stimulating IL-1β and NF-κB signaling. J. Cell Sci. 2015, 128, 3055–3067.
- Wickenhauser, C.; Lorenzen, J.; Thiele, J.; Hillienhof, A.; Jungheim, K.; Schmitz, B.; Hansmann, M.L.; Fischer, R. Secretion of cytokines (interleukins-1 alpha, -3, and -6 and granulocyte-macrophage colony-stimulating factor) by normal human bone marrow megakaryocytes. Blood 1995, 85, 685–691.
- Kaser, A.; Brandacher, G.; Steurer, W.; Kaser, S.; Offner, F.A.; Zoller, H.; Theurl, I.; Widder, W.; Molnar, C.; Ludwiczek, O.; et al. Interleukin-6 stimulates thrombopoiesis through thrombopoietin: Role in inflammatory thrombocytosis. Blood 2001, 98, 2720–2725.
- Behrens, K.; Alexander, W.S. Cytokine control of megakaryopoiesis. Growth Factors 2018, 36, 89–103.
- Wolber, E.M.; Fandrey, J.; Frackowski, U.; Jelkmann, W. Hepatic thrombopoietin mRNA is increased in acute inflammation. Thromb. Haemost. 2001, 86, 1421–1424.
- Burmester, H.; Wolber, E.M.; Freitag, P.; Fandrey, J.; Jelkmann, W. Thrombopoietin production in wild-type and interleukin-6 knockout mice with acute inflammation. J. Interferon Cytokine Res. 2005, 25, 407–413.
- Haas, S.; Hansson, J.; Klimmeck, D.; Loeffler, D.; Velten, L.; Uckelmann, H.; Wurzer, S.; Prendergast, Á.M.; Schnell, A.; Hexel, K.; et al. Inflammation-Induced Emergency Megakaryopoiesis Driven by Hematopoietic Stem Cell-like Megakaryocyte Progenitors. Cell Stem Cell 2015, 17, 422–434.
- Ricciardi, S.; Miluzio, A.; Brina, D.; Clarke, K.; Bonomo, M.; Aiolfi, R.; Guidotti, L.G.; Falciani, F.; Biffo, S. Eukaryotic translation initiation factor 6 is a novel regulator of reactive oxygen species-dependent megakaryocyte maturation. J. Thromb. Haemost. 2015, 13, 2108–2118.
- Nishimura, S.; Nagasaki, M.; Kunishima, S.; Sawaguchi, A.; Sakata, A.; Sakaguchi, H.; Ohmori, T.; Manabe, I.; Italiano, J.E., Jr.; Ryu, T.; et al. IL-1α induces thrombopoiesis through megakaryocyte rupture in response to acute platelet needs. J. Cell Biol. 2015, 209, 453–466.
- Undi, R.B.; Sarvothaman, S.; Narasaiah, K.; Gutti, U.; Gutti, R.K. Toll-like receptor 2 signalling: Significance in megakaryocyte development through wnt signalling cross-talk and cytokine induction. Cytokine 2016, 83, 245–249.
- Tacchini-Cottier, F.; Vesin, C.; Redard, M.; Buurman, W.; Piguet, P.F. Role of TNFR1 and TNFR2 in TNF-induced platelet consumption in mice. J. Immunol. 1998, 160, 6182–6186.
- Beaulieu, L.M.; Lin, E.; Mick, E.; Koupenova, M.; Weinberg, E.O.; Kramer, C.D.; Genco, C.A.; Tanriverdi, K.; Larson, M.G.; Benjamin, E.J.; et al. Interleukin 1 receptor 1 and interleukin 1β regulate megakaryocyte maturation, platelet activation, and transcript profile during inflammation in mice and humans. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 552–564.
- Nakayama, K. Expression of IL-6, IL-6 receptor and its signal transducer gp130 mRNAs in megakaryocytic cell lines. Leuk. Lymphoma 1998, 29, 399–405.
More