Oncolytic virotherapy (OVT) has received significant attention in recent years, especially since the approval of talimogene Laherparepvec (T-VEC) in 2015 by the Food and Drug administration (FDA). Mechanistic studies of oncolytic viruses (OVs) have revealed that most, if not all, OVs induce direct oncolysis and stimulate innate and adaptive anti-tumour immunity. With the advancement of tumour modelling, allowing characterisation of the effects of tumour microenvironment (TME) components and identification of the cellular mechanisms required for cell death (both direct oncolysis and anti-tumour immune responses), it is clear that a “one size fits all” approach is not applicable to all OVs, or indeed the same OV across different tumour types and disease locations. This article will provide an unbiased review of oncolytic reovirus (clinically formulated as pelareorep), including the molecular and cellular requirements for reovirus oncolysis and anti-tumour immunity, reports of pre-clinical efficacy and its overall clinical trajectory. Moreover, as it is now abundantly clear that the true potential of all OVs, including reovirus, will only be reached upon the development of synergistic combination strategies, reovirus combination therapeutics will be discussed, including the limitations and challenges that remain to harness the full potential of this promising therapeutic agent.
- reovirus
- oncolytic virus
- pelareorep
- oncolytic virotherapy
1. Introduction
Advancements in virology and molecular biology techniques over recent decades have allowed us to exploit the anti-tumour potential of oncolytic viruses (OVs) [1]. The unique ability of OVs to exploit oncogenic signalling pathways provides a significant advantage over traditional treatment modalities. OVs are specifically defined as viruses which: (i) preferentially infect and kill malignant cells through viral replication and oncolysis, and (ii) engage the immune system to promote anti-tumour immunity. Additional mechanisms of action have also been reported, including disruption of tumour-associated vasculature or stroma and modulation of the tumour microenvironment (TME)[2][3][4].
An array of OVs—naturally occurring, attenuated, and genetically modified—have been investigated in pre-clinical models and clinical trials but only two have received approval for clinical use: (i) a genetically engineered adenovirus H101, approved in China in 2005 [5], and (ii) the Food and Drug Administration (FDA)-approved talimogene laherparepvec (T-VEC)—a herpes simplex virus type 1 (HSV-1) genetically engineered to limit neurovirulence and promote an immunostimulatory environment[6][7].
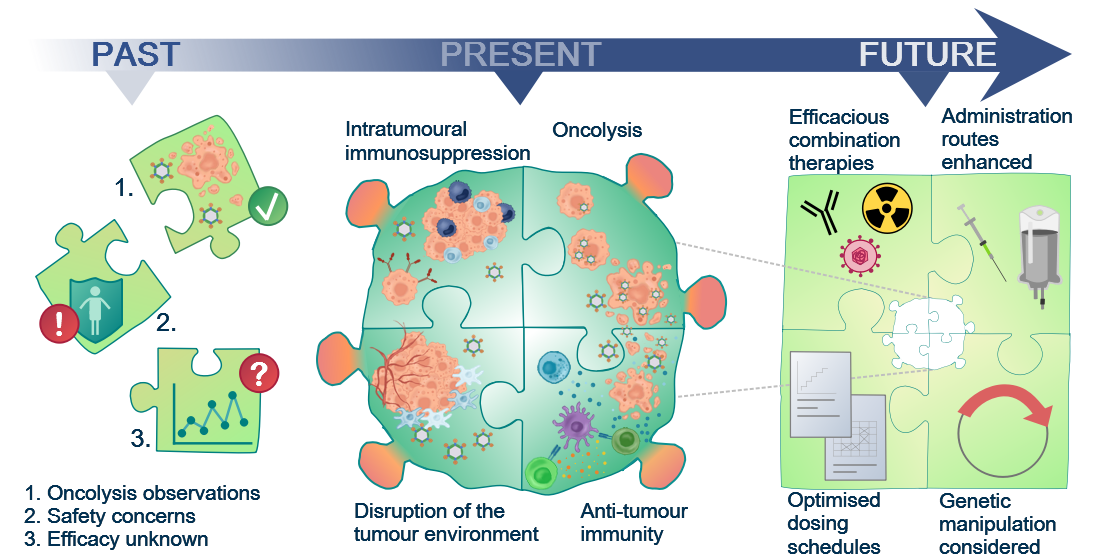
Figure 1. Although the oncolytic capacity of various viruses was noted already in the 1950s, development of oncolytic viruses (OVs) as therapeutic agents was limited by safety and efficacy concerns for a long time. With a greater understanding of virology and molecular techniques, the interest in OVs has peaked in recent years and a number of viruses, including reovirus, are now in clinical development. Today, we know that OVs exert their anti-tumour effects in several ways, e.g. direct oncolytic killing through replication in malignantly transformed cells; innate and adaptive anti-tumour immunity; disruption of the local tumour microenvironment sustaining the tumour; and intratumoural immunosuppressive effects, e.g. recruitment of regulatory T cells and enhanced expression of immune checkpoints on tumour cells. This knowledge will allow future initiatives to the identify the most efficacious combination therapies, optimised dosing schedules, and administration routes required for the success of reovirus as a therapeutic agent.
2. The Emergence of Reovirus as a Therapeutic Agent
The Reoviridae family of viruses has found hosts in mammals, fish, birds and plants [8][9]. Three serotypes of mammalian orthoreovirus have been identified: type one Lang, type two Jones, and type three Abney and Dearing [10]. Each differs in its in vivo tropism, despite a high degree of genetic similarity [11]. Type-specific diversity occurs in the S1 gene, encoding the outer capsid σ1 attachment protein, which has undergone significant evolutionary divergence[12]. Orthoreovirus type two Jones was the first serotype observed to replicate specifically in malignant cell lines[13]; however, it is the mammalian orthoreovirus type three Dearing strain (T3D)—now manufactured as pelareorep but previously known as Reolysin®—that has made progress as a therapeutic agent. Mammalian orthoreovirus T3D (hereafter referred to as reovirus) is typically isolated from human gastrointestinal and upper respiratory tracts[14][15]. In most individuals, infection proceeds asymptomatically causing mild enteric or respiratory illness in young children and being relatively non-pathogenic in adults, in line with its designation as a respiratory enteric orphan virus (reovirus)[10]. There have been sporadic reports of severe pathology associated with reovirus infection in infants and immunocompromised individuals [9][16][17][18][19][20][21] and more recently, reovirus has been implicated in coeliac disease by promoting a TH1 immune response, a response that bodes well for its use as an immunotherapeutic tool although oral delivery should be avoided to limit these potential unwanted side effects[22].
Reovirus is a non-enveloped, double-stranded (ds) RNA virus approximately 85 nm in diameter, with two concentric icosahedral protein capsids[23]. The outer and inner capsids protect the dsRNA genome which comprises 23.5 kbp in ten segments termed large (L1-3), medium (M1-3), or small (S1-4) according to size [23][24][25]. The gene segments encode eight structural proteins (λ1-3, µ1-2, and σ1-3) and the non-structural proteins, µNS and σNS[26]. μ1 and σ3 form part of the outer capsid, λ3 forms a subunit of the RNA polymerase and σ1 and λ2 are important for viral attachment, although σ1 initiates target cell entry[23]. The proteins also protect the virus from immune-surveillance by preventing a host anti-viral interferon (IFN) response; σ3 binds to dsRNA and prevents its binding to dsRNA-dependent protein kinase R (PKR; a dsRNA sensor)[27] and μNS sequesters the IFN transcription factor (interferon regulatory factor 3; IRF3) and inhibits its translocation to the nucleus[28].
3. Tumour Specificity and Replication
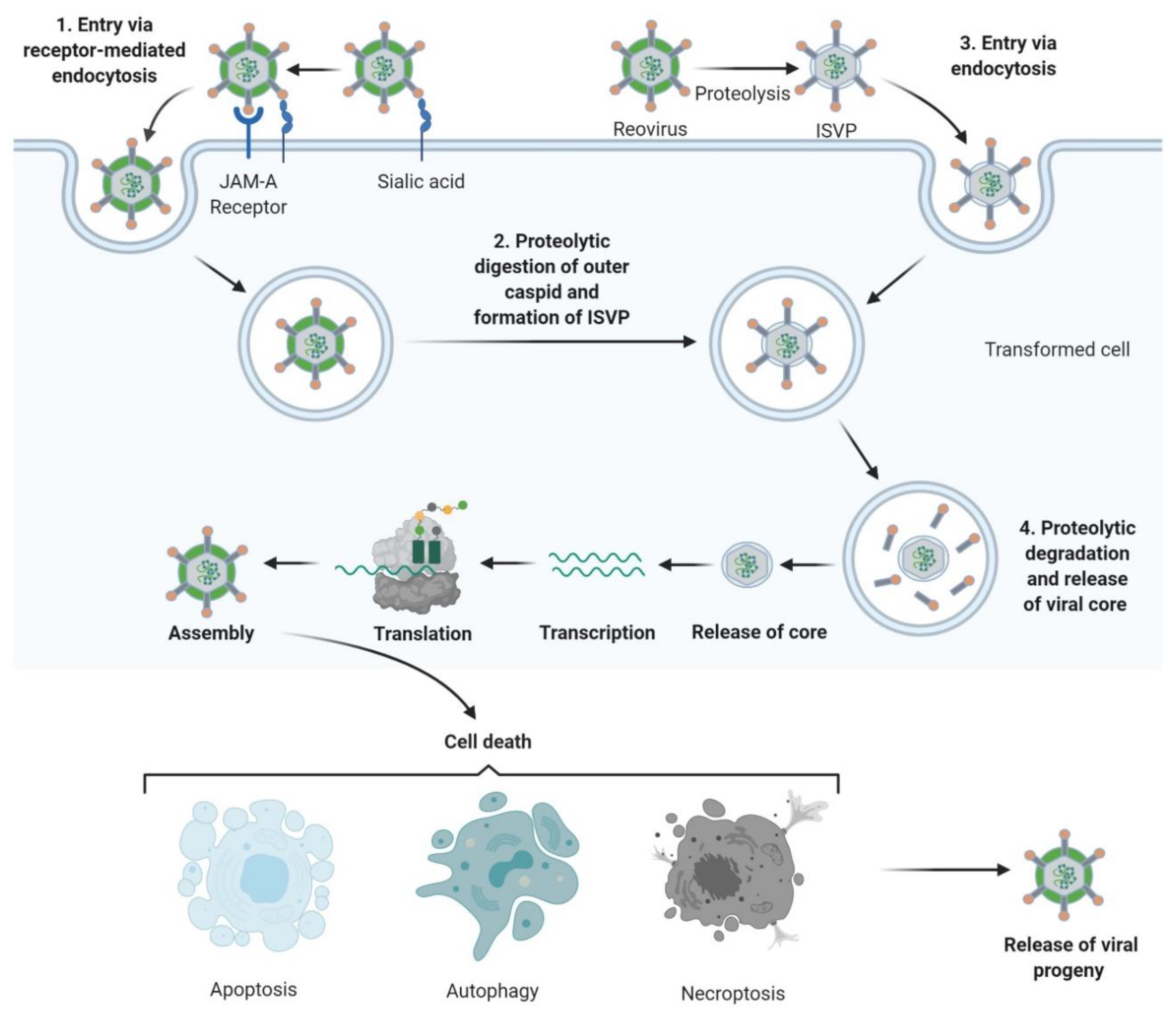
The reovirus life-cycle is shown in Figure 2. Viral entry occurs over multiple steps, the first being a low-affinity “tethering” of the reovirus σ1 protein to cell surface sialic acid[29][30]. Subsequently, σ1 engages junctional adhesion molecule A (JAM-A), the canonical reovirus receptor [31][32][33], which is ubiquitously expressed throughout the body and has several important roles in normal cellular processes including tight junction formation, leukocyte migration, and angiogenesis [34]. Fortuitously, JAM-A is also overexpressed in several cancers, including both haematological and solid malignancies[35][36][37][38][39][40][41]. Following reovirus engagement with JAM-A and receptor-mediated endocytosis, the viral particle undergoes acid-dependent cathepsin-mediated proteolysis within the endosome[42][43]to form an intermediate subviral particle (ISVP) characterised by the loss of σ3 and cleavage of µ1 [44]. The proteolytic uncoating, principally by cathepsins L and B, is critical for penetration of the endosome membrane by µ1; ISVPs undergo a conformational change causing autocleavage of µ1 into µ1N which triggers pore formation in the endocytic membrane[45] and delivers transcriptionally active reovirus into the cytosol[46][47] for replication. Capped, positive-sense single stranded (ss) RNA serves as mRNA for protein translation and provides a template for replication of nascent dsRNA genomes [48]. Transcription and translation occur in cytoplasmic “viral factories”[49][50], with packaging of the segmented genome into virions occurring concomitantly with RNA synthesis[51][52]. Viral egress can be non-cytolytic in the absence of transformation; however, the release of progeny virus is typically lytic in permissive, transformed cells[53][54].
Figure 2. Reovirus replication: 1. Reovirus is first tethered via a weak interaction between σ1 and cell surface sialic acid; σ1 then binds with high affinity to junctional adhesion molecule A (JAM-A) resulting in internalization of the virus via receptor-mediated endocytosis. 2. Once internalized, the virus is transported to early and late endosomes where it undergoes proteolytic digestion to remove the outer capsid protein σ3 resulting in the formation of infectious subvirion particles (ISVPs). 3. Alternatively, ISVPs may be formed by extracellular proteases within the tumour environment allowing direct entry into cells via membrane penetration. 4. After further proteolytic degradation a transcriptionally active viral core is released into the cytoplasm. Transcription and translation occur ultimately leading to the assembly of new viral progeny, host cell death and progeny release. Figure created using Biorender (https://biorender.com/.
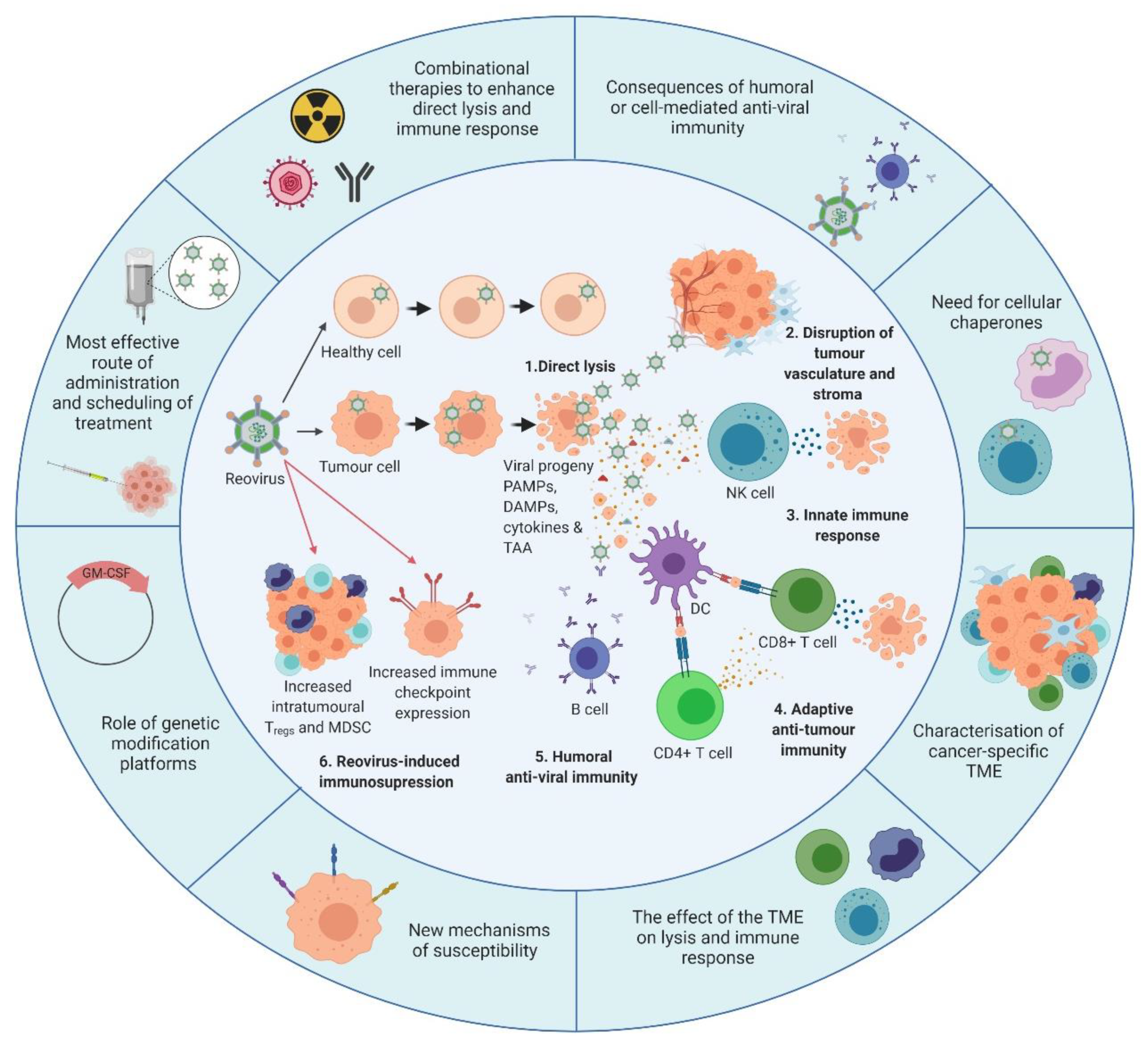
Figure 3. Overview of reovirus mechanisms of action and the developments required. The inner circle illustrates what is currently known about reovirus. 1. In healthy cells, anti-viral immune responses limit reovirus replication and prevent lytic killing. By contrast, oncogenic signalling pathways render tumour cells susceptible to reovirus replication and direct oncolysis. 2. Reovirus replicates in the tumour vasculature and stroma due to reciprocal cell:cell interactions which alter anti-viral signalling. 3. Infection of tumour cells leads to the release of viral progeny, cytokines and tumour-associated antigens (TAAs), which initiates innate anti-tumour immunity including cytokine-mediated bystander killing and natural killer (NK) cell-mediated cytotoxicity. 4. Adaptive anti-tumour immunity is generated following the phagocytosis of TAAs by dendritic cells (DCs) and presentation of TAAs to CD4+ve and CD8+ve T cells, which facilitates priming of tumour-specific cytotoxic T lymphocytes (CTLs). 5. In addition to innate and adaptive anti-tumour immune responses, humoral anti-viral immunity is induced, leading to the production of reovirus-specific neutralising antibodies (NAbs). 6. Following induction of anti-tumour/anti-viral immune responses, regulatory immune mechanisms are “switched-on” to control ongoing immune responses, including upregulation of immune checkpoints and increased levels of regulatory T cells (Tregs) and/or myeloid-derived suppressor cells (MDSCs). The outer circle highlights priority research areas to improve reovirus efficacy. These include gaining a greater understanding of: (i) the consequence of humoral and/or cell-mediated anti-viral immunity on reovirus efficacy which would inform the development of, or the requirement for, cellular chaperones; (ii) the tumour microenvironment (TME) and how it influences reovirus oncolysis and anti-tumour immunity; (iii) the cellular determinants utilized by reovirus for direct oncolysis, including mechanisms of reovirus resistance; (iv) the potential benefits of genetically-modified reovirus platforms; (v) reovirus scheduling to maximize virus delivery and efficacy including the best route of virus administration; and (vi) combinatorial approaches that are designed to boost both direct oncolysis and anti-tumour immune responses. PAMPs: pathogen-associated molecular patterns; DAMPs: damage-associated molecular patterns; GM-CSF: granulocyte-macrophage colony stimulating factor. Figure created using Biorender.com)