The recycling of sodium borohydride poses a huge challenge to the drive towards a hydrogen economy. Mechano-chemical, thermo-chemical and electrochemical are the only reported methods of recycling sodium metaborate into sodium borohydride. Much attention have been devoted towards the mechano-chemical and thermo-chemical methods of reduction, but little focus is devoted to electrochemical methods. This mini-review describes the electrochemical behaviour of borohydride (BH
The recycling of sodium borohydride poses a huge challenge to the drive towards a hydrogen economy. Mechano-chemical, thermo-chemical and electrochemical are the only reported methods of recycling sodium metaborate into sodium borohydride. Much attention have been devoted towards the mechano-chemical and thermo-chemical methods of reduction, little focus is devoted to electrochemical methods. This research describes the electrochemical behaviour of borohydride (BH
4
ˉ) and metaborate (BO
2
ˉ) anion in alkaline solutions. The electrochemical characteristics of BH
4
ˉ is controlled by the alkaline concentration, the concentration of the BH
4
ˉ and the type of electrode material. The attempts to electro-reduce the BO
2
ˉ into BH
4
ˉ is reviewed and the challenges, suggestions and future outlook of electro-reduction to recycle the BO
2
ˉ into BH
4
ˉ is highlighted.
- sodium metaborate
- sodium borohydride
- electro-reduction
[1]1. Introduction
2. Electrolysis of BO2− into BH4−
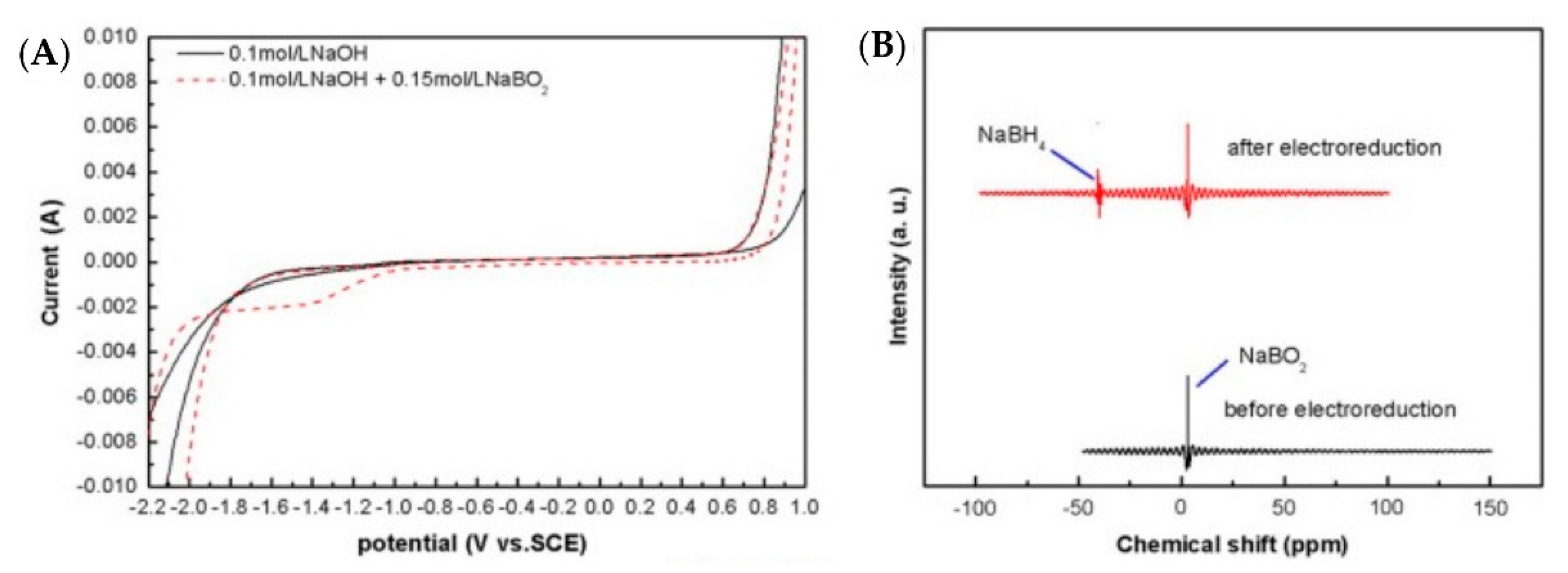
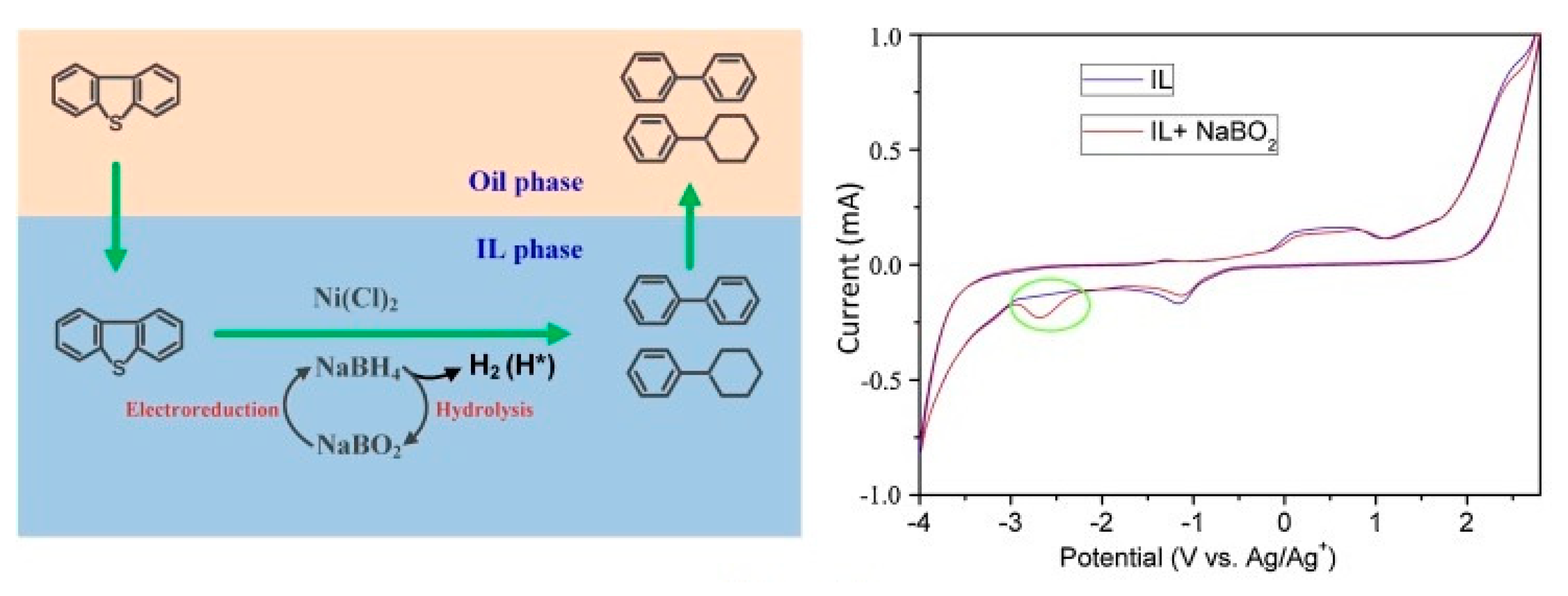
A few patents describe the electro-reduction of BO2− into BH4− in an aqueous medium [49,50[21][22][23],51], but later reports [26][24] did not verify any viable conversion of BO2− into BH4− in an aqueous medium, although the current efficiencies of the conversion, 20–25%, were reported in those patents [49,50,51][21][22][23]. This could be due to the iodometric titration, which yields higher NaBH4 formation, which was adopted by the patents [26][24]. McLafferty et al. [52][25] also performed experiments to verify several patents [52,53,54][25][26][27] that claim the successful electrochemical reduction of BO2− into BH4−. They performed electrolysis of boric acid on mercury pool cathode in a divided electrolysis cell with cationic exchange membrane with sulfuric acid as the anolyte and BO2− in 2 M NaOH as the catholyte. Since mercury has high overpotential for the hydrogen evolution reaction (HER), it was selected as the cathode in this study, and the potential of the mercury cathode was maintained between −2.16 and −2.60 V (vs. Hg/HgO). The authors found no presence of the BH4− using cyclic voltammetry of alkaline solution even after two days of electrolysis. The same results were obtained with the hydrolysis of BO2− in the catholyte at gold and Cu electrodes, to reproduce the work of Jiang et al. [55][28], but no traces of the BH4− were detected using CV of the catholyte after electrolysis. Gold cathode was used again in pulsed potential electrolysis and there was no presence of any BH4− detected using CV. Titanium, Ag and Pb, which possess high overpotentials of HER, also did not show any conversion into BH4− in the alkaline reduction of the BO2− in the divided electrolysis cell. The authors used tetraalkylammonium hydroxide (TAAH) to increase the overpotential of the HER. All experiments concluded that the TAA+ cation was ineffective in absorbing at the cathode surface due to the replacement of water molecules at higher negative potentials, which promoted the HER. The authors also tried to modify the cathode surface using a chelating compound or a cationic polyelectrolyte to coat the gold electrode surface, forming a self-assembled monolayer (SAM), which promotes the attraction of the BO2− at small distances from the gold electrode surface. The chelating agent utilised for this purpose was 1-thioglycerol, which consists of a thiol group, which strongly bounds to the gold surface, and a diol group, which could bind with the BO2−, while a quaternized poly(4-vinylpyridine) was selected as the cationic polyelectrolyte. The electrolysis was repeated as with the high overpotential metal cathode, but no presence of the BH4− ion was detected in the CV. Sanli et al. [56][29] carried out electrolyses in 1 M NaOH + 0.1 M NaBO2 at 0.5 V for 24 h and 48 h with Ag gauze cathode at room temperature and atmospheric pressure in an undivided electrochemical cell. The authors performed iodometric titration for the determination of the conversion efficiency of sodium metaborate into sodium borohydride. It was observed that there was 10% conversion after 24 h of electrolysis, reaching 17% (by iodometric titration) after 48 h. It was obvious that the low conversion efficiency was due to the HER at the cathode, which is a strong competing reaction with the electro-reduction of metaborate into borohydride. The hydrogen is produced in situ at the cathode: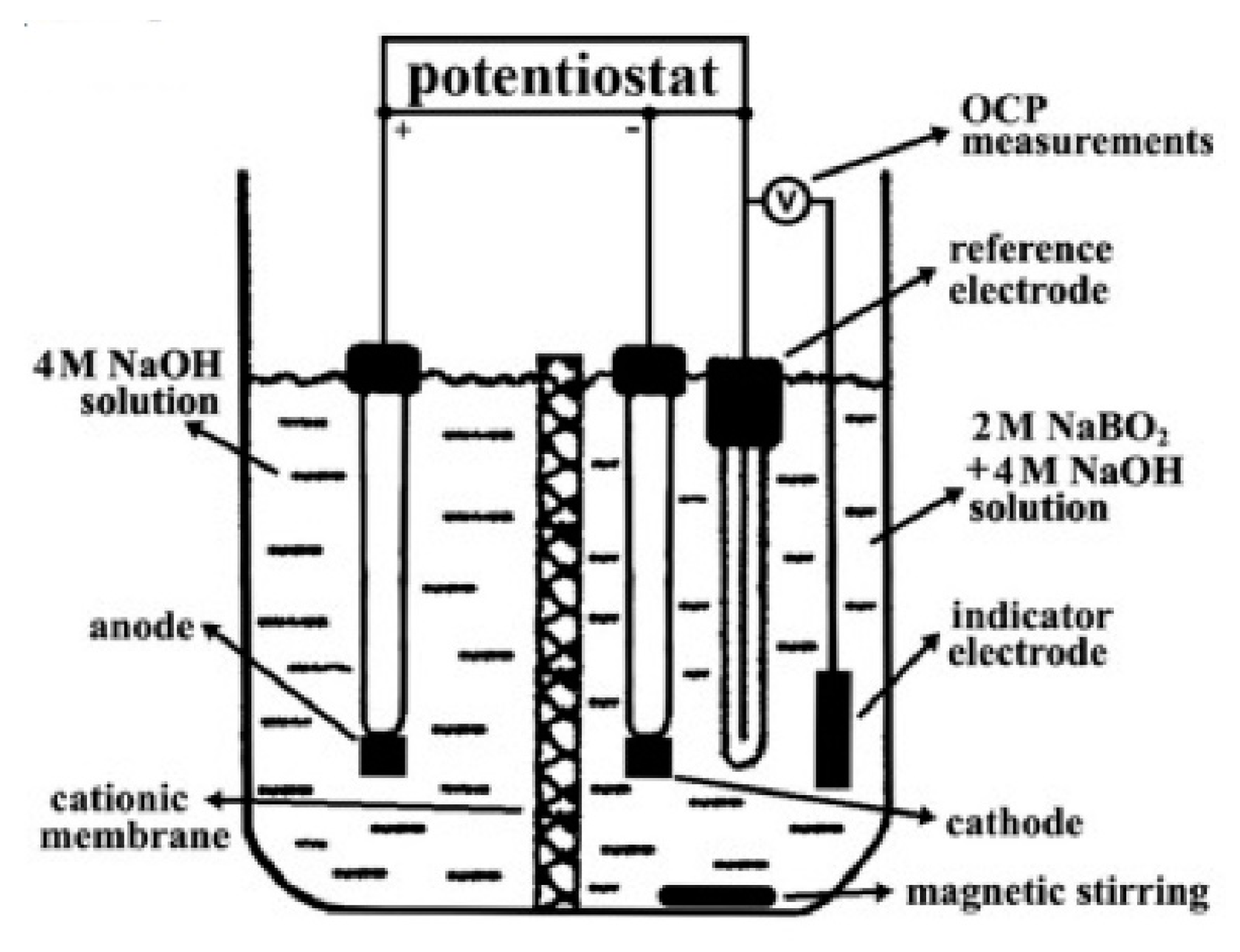
References
- Wan Jefrey Basirun, Syed Tawab Shah, Md. Shalauddin, Shamima Akhter, Nazzatush Shimar Jamaludin, Adeeb Hayyan; A Review of Electrochemical Reduction of Sodium Metaborate. Energies 2023, 16, 15, https://doi.org/10.3390/en16010015.Schlesinger, H.; Brown, H.C.; Finholt, A. The preparation of sodium borohydride by the high temperature reaction of sodium hydride with borate esters. J. Am. Chem. Soc. 1953, 75, 205–209.
- Demirci, Ü.B. Sodium borohydride for the near-future energy: A “rough diamond’’ for Turkey. Turk. J. Chem. 2018, 42, 193–220.
- Kojima, Y.; Haga, T. Recycling process of sodium metaborate to sodium borohydride. Int. J. Hydrogen Energy 2003, 28, 989–993.
- Li, Z.; Liu, B.; Zhu, J.; Morigasaki, N.; Suda, S. NaBH4 formation mechanism by reaction of sodium borate with Mg and H2. J. Alloys Compd. 2007, 437, 311–316.
- Liu, B.H.; Li, Z.P.; Zhu, J.K.; Morigasaki, N.; Suda, S. Sodium borohydride synthesis by reaction of Na2O contained sodium borate with al and hydrogen. Energy Fuels 2007, 21, 1707–1711.
- Eom, K.; Cho, E.; Kim, M.; Oh, S.; Nam, S.-W.; Kwon, H. Thermochemical production of sodium borohydride from sodium metaborate in a scaled-up reactor. Int. J. Hydrogen Energy 2013, 38, 2804–2809.
- Figen, A.K.; Pişkin, S. Microwave assisted green chemistry approach of sodium metaborate dihydrate (NabO2·2H2O) synthesis and use as raw material for sodium borohydride (NaBH4) thermochemical production. Int. J. Hydrogen Energy 2013, 38, 3702–3709.
- Ou, T.; Panizza, M.; Barbucci, A. Thermochemical recycling of hydrolyzed NaBH4. Part II: Systematical study of parameters dependencies. Int. J. Hydrogen Energy 2013, 38, 15940–15945.
- Hsueh, C.-L.; Liu, C.-H.; Chen, B.-H.; Chen, C.-Y.; Kuo, Y.-C.; Hwang, K.-J.; Ku, J.-R. Regeneration of spent-NaBH4 back to NaBH4 by using high-energy ball milling. Int. J. Hydrogen Energy 2009, 34, 1717–1725.
- Kong, L.; Cui, X.; Jin, H.; Wu, J.; Du, H.; Xiong, T. Mechanochemical synthesis of sodium borohydride by recycling sodium metaborate. Energy Fuels 2009, 23, 5049–5054.
- Liu, C.-H.; Kuo, Y.-C.; Chen, B.-H.; Hsueh, C.-L.; Hwang, K.-J.; Ku, J.-R.; Tsau, F.; Jeng, M.-S. Synthesis of solid-state NaBH4/Co-based catalyst composite for hydrogen storage through a high-energy ball-milling process. Int. J. Hydrogen Energy 2010, 35, 4027–4040.
- Çakanyıldırım, Ç.; Gürü, M. Processing of NaBH4 from NaBO2 with MgH2 by ball milling and usage as hydrogen carrier. Renew. Energy 2010, 35, 1895–1899.
- Chen, W.; Ouyang, L.; Liu, J.; Yao, X.; Wang, H.; Liu, Z.; Zhu, M. Hydrolysis and regeneration of sodium borohydride (NaBH4)—A combination of hydrogen production and storage. J. Power Sources 2017, 359, 400–407.
- Lang, C.; Jia, Y.; Liu, J.; Wang, H.; Ouyang, L.; Zhu, M.; Yao, X. NaBH4 regeneration from NABO2 by high-energy ball milling and its plausible mechanism. Int. J. Hydrogen Energy 2017, 42, 13127–13135.
- Zhong, H.; Ouyang, L.Z.; Ye, J.S.; Liu, J.W.; Wang, H.; Yao, X.D.; Zhu, M. An one-step approach towards hydrogen production and storage through regeneration of NaBH4. Energy Storage Mater. 2017, 7, 222–228.
- Zhong, H.; Ouyang, L.; Zeng, M.; Liu, J.; Wang, H.; Shao, H.; Felderhoff, M.; Zhu, M. Realizing facile regeneration of spent NaBH4 with Mg–Al alloy. J. Mater. Chem. A 2019, 7, 10723–10728.
- Qin, C.; Ouyang, L.; Wang, H.; Liu, J.; Shao, H.; Zhu, M. Regulation of high-efficient regeneration of sodium borohydride by magnesium-aluminum alloy. Int. J. Hydrogen Energy 2019, 44, 29108–29115.
- Le, T.T.; Pistidda, C.; Puszkiel, J.; Milanese, C.; Garroni, S.; Emmler, T.; Capurso, G.; Gizer, G.; Klassen, T.; Dornheim, M. Efficient synthesis of alkali borohydrides from mechanochemical reduction of borates using magnesium–aluminum-based waste. Metals 2019, 9, 1061.
- Zhu, Y.; Ouyang, L.; Zhong, H.; Liu, J.; Wang, H.; Shao, H.; Huang, Z.; Zhu, M. Closing the loop for hydrogen storage: Facile regeneration of NaBH4 from its hydrolytic product. Angew. Chem. Int. Ed. 2020, 132, 8701–8707.
- Nunes, H.X.; Silva, D.L.; Rangel, C.M.; Pinto, A.M. Rehydrogenation of sodium borates to close the NaBH4-H2 cycle: A review. Energies 2021, 14, 3567.
- Cooper, H. Electrolytic Process for the Production of Alkali Metal Borohydrides. U.S. Patent 3734842, 22 May 1973.
- Sharifian, H.; Dutcher, J.S. Production of Quaternary Ammonium and Quaternary Phosphonium Borohydrides. U.S. Patent 3734842, 27 February 1990.
- Hale, C.H.; Sharifian, H. Production of Metal Borohydrides and Organic Onium Borohydrides. U.S. Patent 4931154, 5 June 1990.
- Gyenge, E.; Oloman, C. Electrosynthesis attempts of tetrahydridoborates. J. Appl. Electrochem. 1998, 28, 1147–1151.
- McLafferty, J.; Colominas, S.; Macdonald, D. Attempts to cathodically reduce boron oxides to borohydride in aqueous solution. Electrochim. Acta 2010, 56, 108–114.
- Amendola, S. Electroconversion Cell. U.S. Patent 6,497,973 B1, 24 December 2002.
- Kawai, M.; Ito, M. Japanese Patent 2003-247088, 2003.
- Wang, J.; Sun, Y.; Liang, Z. Preliminary study on electrochemical reduction of sodium metaborate to produce sodium borohydride. J. Taiyuan Univ. Tech. 2006, 37, 539.
- Sanli, A.E.; Kayacan, İ.; Uysal, B.Z.; Aksu, M.L. Recovery of borohydride from metaborate solution using a silver catalyst for application of direct rechargable borohydride/peroxide fuel cells. J. Power Sources 2010, 195, 2604–2607.
- Santos, D.; Sequeira, C. On the electrosynthesis of sodium borohydride. Int. J. Hydrogen Energy 2010, 35, 9851–9861.
- Shen, Y.; Sun, T.; Jia, J. A novel desulphurization process of coal water slurry via sodium metaborate electroreduction in the alkaline system. Fuel 2012, 96, 250–256.
- Zhu, Q.-Y.; Zhu, C.-G.; Wang, F.-W.; Wei, Y.-J. Preparation of sodium borohydride by copper electrolysis. Asian J. Chem. 2013, 25, 7749–7752.
- Shu, C.; Sun, T.; Jia, J.; Lou, Z. A novel desulfurization process of gasoline via sodium metaborate electroreduction with pulse voltage using a boron-doped diamond thin film electrode. Fuel 2013, 113, 187–195.
- Shu, C.; Lai, F.; Zhu, F.; Luo, D. Optimal extractive and reductive desulfurization process using sodium borohydride in situ generated via sodium metaborate electroreduction in ionic liquid. Chem. Eng. Process.-Process Intensif. 2020, 150, 107869.