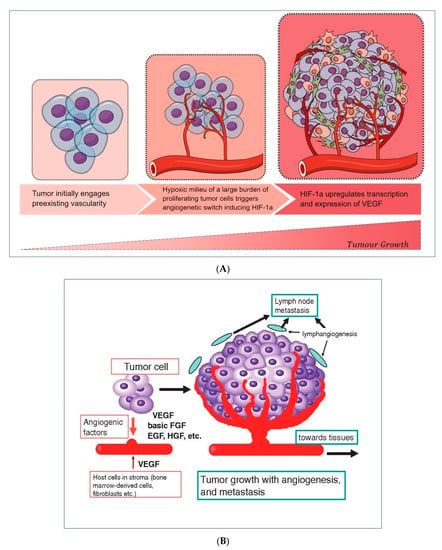
Figure 2. (
A) Hypoxic environment stimulates neoangiogenesis, thanks to a circuit involving HIF-1α -induced VEGF release by tumor cells. (
B) A wide range of cellular released growth factors, derived from a tumor, stroma, and leukocytes, ensures the formation of new vascularity and the sustainment of cell growth, allowing the worsening progression of the initial tumor burden.
Patients with HCC showed a significant increase in VEGF after anticancer therapy compared to the values reported at the time of diagnosis, as well as to the levels of lymphocytes
[9]. This may be partly explained by the rebound effect of VEGF, induced by hypoxia following locoregional treatments, often associated with treatment failure and low survival rates in patients
[10]. Levels of VEGF are increased in patients who later experienced progression of HCC compared to those who remained stable
[11]. Additionally, higher VEGF levels prior to sorafenib treatment (a multikinase inhibitor employed in several locally recurrent or metastatic solid tumors, including HCC) are associated with shorter survival
[12][13][12,13].
The Tyrosine kinase proteins with Ig and EGF-homology domains 1 and 2 (Tie1 and Tie2) and their angiopoietin ligands 1–4 (Ang1, 2, 3, and 4) play a key role during the late phase of angiogenesis and are responsible for the maturation of newly established vascular structures. Ang1 and Ang2 have been deeply studied and characterized. The activity of the Angiopoietin/Tie system determines the stabilization of new vessels
[14][25]. There is growing evidence that the angiopoietin/Tie signal can modify ongoing inflammation
[15][14]. Ang1 appears to be a powerful activator of Tie2, as well as a regulator of blood vessel formation and development. Experimental studies showed that Ang1 acts as an anti-inflammatory molecule
[16][26] but can induce pulmonary hypertension as a complication
[17][27]. Ang1 also neutralizes tissue factor activity that is relevant for the induction of coagulation, thrombosis, and inflammatory response. Furthermore, Ang1 reduces the adhesion of VEGF-related leukocytes to the endothelium
[18][19][28,29]. Conversely, Ang2 acts as a competitive antagonist of Ang1, deregulates the signal pathway of Tie2
[14][25], and plays a pro-inflammatory role
[20][21][30,31]. Additionally, significantly high Ang2 serum levels have been observed in patients during liver carcinogenesis
[22][32]. An increase in Ang2 levels has been observed in correlation with the liver disease progression
[9][23][9,33], while Ang1 negatively correlates with the model for end-stage liver disease (MELD) and the hepatic fibrosis index. All these data confirm the potential diagnostic utility of Ang1 and Ang2 levels as developing new quantitative biomarkers for staging cirrhosis. Serum Ang2 concentrations decrease significantly after treatment with Direct Antiviral Drugs (DAAs), and, consequently, the Ang2/Ang1 ratio also drops. Ang2 is potentially useful in monitoring antiviral therapy
[9].
It has been shown that the Hepatocyte growth factor (HGF) is over-expressed in HCC compared to the normal and cirrhotic liver without signs of neoplasia
[9][24][25][9,15,34]. Expression of HGF and its receptor supports the existence of both autocrine and paracrine mechanisms of HGF action in HCC if compared to the unique paracrine mechanism found in normal liver tissue (in the absence of cancer), suggesting that it also plays a role in tumor development and progression
[24][25][26][15,34,35]. Stellate cells and myofibroblasts are induced to secrete HGF from tumor cell products, and HGF, in turn, stimulates tumor cell invasiveness. Recent reports show that higher serum HGF levels negatively correlate with patient survival time
[27][36] and positively with tumor size
[28][37].
Furthermore, the comparison of cirrhotic patients with and without HCC suggests that HGF levels are potentially useful for monitoring the onset of HCC after a diagnosis of cirrhosis
[9]. Interestingly, patients with lower HGF levels prior to treatment display major benefits from sorafenib therapy in terms of overall survival and time to progression
[12].
Platelet endothelial cell adhesion molecule-1 (PECAM-1), also known as CD31, is normally found on the surface of endothelial cells, platelets, leukocyte subpopulations, and Kupffer cells
[29][38]. This molecule is highly expressed within the vascular compartment but largely concentrated at junctions between adjacent cells, and its receptors mediate these interactions that play a crucial role during angiogenesis. In this context, PECAM-1 can mediate both homophilic and heterophilic adhesion
[30][16]. PECAM-1 has been found to positively correlate with MELD, and its identification may aid in assessing the degree of tumor angiogenesis, which may indicate a rapidly growing tumor
[31][39]. Furthermore, PECAM-1 promotes the formation of metastases by inducing the epithelium-mesenchymal transition in HCC by increasing the regulation of β1 integrin through the FAK (focal adhesion kinase) /Akt signaling pathway
[32][40].
2.2. Stimulators of Chronic Inflammation, Liver Fibrosis, and Proliferation
Interleukin (IL)-6 acts as an important inducer of the acute phase response and infection defense in the liver
[33][41]. IL-6 binds to the signal-transducing subunit gp130 on target cells either in complex with the membrane-bound or with the soluble IL-6 receptor to activate intracellular signaling. By the latter ‘trans-signaling’ mechanism, IL-6 can target monocyte chemotaxis and maintain sustained chronic inflammation towards any injured tissue
[34][35][42,43]. Increased serum levels of IL-6 have been found in patients with advanced HCC compared to those with the early stage
[36][44]. Additionally, elevated serum IL-6 levels in HCC patients undergoing hepatectomy are associated with lower overall survival and are prone to early relapses
[37][45]. Research results from mouse models of HCC have shown that isolated HCC progenitor cells can give rise to cancer in the presence of ongoing liver damage and that these cells promote their own growth and progress to malignancy via autocrine IL-6 signaling
[38][17]. A clinical study analyzing 128 HCC patients treated with sorafenib evaluated the prognostic value of serum IL-6 levels before treatment. Elevated pretreatment IL-6 concentrations have been found to be an independent predictor of poor overall survival, although there is no association with the efficacy of sorafenib
[39][18].
Transforming growth factors (TGF)-α and TGF-β are closely related to the hepatocarcinogenesis process. Normal hepatocytes show low TGF-α expression compared to tumor cells. In fact, following chronic inflammation due to persistent liver damage, the secreted cytokine pool upregulates TGF-α in the liver and, consequently, regeneration, proliferation, hepatocyte dysplasia, and, ultimately, the development of HCC
[40][46]. TGF-β is a key regulator of the late phase of inflammatory processes, not only promoting tissue repair but also inhibiting leukocyte activation and infiltration, acting at least in part using control of adhesion molecules on parenchymal cells
[41][47]. In this way, TGF-β counteracts the effects of proinflammatory cytokines, leading to the inhibition of cellular processes, such as proliferation, differentiation, and survival. Paradoxically, cancer cells may exploit these microenvironment modifications to their advantage
[42][48]. During carcinogenesis, malignant cells can often attenuate the suppressive TGF-β signaling by altering the expression of its receptors but also hijacking the signaling cascade. HCC cell lines with metastatic potential have been described to downregulate TGF-βR2. Interestingly, a reduced expression of TGF-βR2 in HCC correlates with larger tumor size and various metastatic features, such as poor differentiation, portal vein invasion, and intrahepatic metastases
[43][44][49,50]. In the early stages of cancer, TGF-β acts as a tumor suppressor by inducing cytostasis and apoptosis, while in the later stages, it promotes pro-tumorigenic events, such as the transition of epithelial cells to mesenchymal, invasion, metastasis, and angiogenesis
[45][51]. The simultaneous exposure (or addition) of TGF-β and IL-6 to human HCC cell cultures (Huh) highlighted an attenuation of the pro-proliferative effects induced by IL-6 by TGF-β. This explains a decrease in the transcription levels of the IL-6 receptor (IL-6R), in the expression of STAT-3 (signal transducer and activator of transcription) induced by IL-6, its nuclear localization, and, finally, a reduced activation of p65 compared to the unperturbed activation of the pathway. SMAD (small mother against decapentaplegic)-dependent TGF-β, resulting in the transition from epithelial to mesenchymal cells and, thus, loss of cell polarity and cell adhesion, as well as the acquisition of invasive and migratory properties, coupled with cell growth arrest
[46][52].
IL-10 is a potent anti-inflammatory cytokine. Its role in HCC is less documented than in viral infections. A recent meta-analysis showed that IL-10 levels in HCC patients increased compared to cirrhotic patients and healthy controls but not compared to viral hepatitis patients
[47][53]. Individuals with resectable HCC and IL-10 levels > 12 pg/mL display worse postoperative outcomes
[48][54]. A study showed that in unresectable HCC, the serum levels of IL-10 acted as a negative prognostic factor
[49][19].
Tumor necrosis factor (TNF) is a cytokine produced by proteolytic cleavage from a transmembrane protein precursor (mTNF) into a soluble TNF (sTNF). sTNF binds TNF receptors (TNFR1) (constitutively expressed in most tissues) and TNFR2 (expressed only in hematopoietic and endothelial cells), while mTNF only the type 2 receptor. TNF induces numerous biological responses in the liver, such as apoptosis, necrosis hepatocytes, hepatic inflammation, and regeneration, as well as the progression of HCC
[50][23].
Expression of IL-6 and TNF-α during chronic liver injury activates the transduction pathway downstream of the transcription factor STAT3, which drives neoplastic transformation in the hepatic microenvironment
[51][62]. IL-6 mediates its pro-proliferative effects through activation and direct interaction with the p65 subunit of NF-kB, activation of which is associated with a frequent and early event in liver fibrosis and HCC, regardless of etiology
[52][53][63,64].
2.3. Liver Tumor Inducers
IL-16 is a pleiotropic cytokine whose activity influences both the chemical attraction and the modulation of the activation of T lymphocytes
[54][20]. It has been identified as an important over-expressed cytokine in human liver tissue of HCC in both non-tumor and tumor regions compared to benign tumors and non-cancerous liver levels. Furthermore, IL-16 production can activate the ERK (extracellular signal-regulated kinase)/cyclin D1 signaling pathway, leading to tumor growth
[55][65].
Different research groups evaluate osteopontin as an early marker of HCC. Produced by Kupffer cells, stellate cells, and hepatocytes, this cytokine is highly expressed at sites of inflammation and tissue remodeling
[56][66]. Osteopontin mediates a wide range of biological functions in the immune and vascular systems and has been extensively studied in numerous cancers
[57][24]. An increase in serum osteopontin levels was found in individuals with HCC compared to liver cirrhosis alone or chronic liver disease. The specific diagnostic efficacy of osteopontin in detecting early-stage HCC by differentiating them from non-HCC patients varies considerably among studies. Two studies report that osteopontin levels within two years of diagnosis have a reasonable predictive value of HCC with an AUC (area under the curve) of 0.82
[58][59][67,68].
After HCV eradication, patients undergo follow-up for the risk of developing HCC. Growth differentiation factor 15 (GDF15) is a cytokine, induced by mitochondrial dysfunction or oxidative stress. In one study, serum levels of GDF15 were measured from patients with chronic HCV infection without a history of HCC who had achieved a sustained virological response with DAAs. Serum levels of GDF15 were higher in patients with HCC onset after treatment with DAAs than in untreated patients. Furthermore, the score obtained using an algorithm composed of the GDF15, AFP (alpha-fetoprotein), and the FIB-4 index stratifies the risk of developing HCC de novo after the elimination of HCV
[60][22].
3. Detection and Measurement of Cytokines
The demand for increased testing, particularly for early events of hepatocellular carcinogenesis, for its recurrence or detection of minimal residual disease in those asymptomatic patients requiring alternative approaches. Different methods for measurement of cytokines are currently available, including immunoassays for the detection of single molecules (ELISA, western blot), multiplex assays (chemiluminescent, bead-based (Luminex), and planar antibody arrays), and mass spectrometry
[61][69].