Acridine derivatives are a class of compounds that are being extensively researched as potential anti-cancer drugs. Acridines are well-known for their high cytotoxic activity; however, their clinical application is restricted or even excluded as a result of side effects. The photocytotoxicity of propyl acridine acts against leukaemia cell lines, with C1748 being a promising anti-tumour drug against UDP-UGT’s. CK0403 is reported in breast cancer treatment and is more potent than CK0402 against estrogen receptor-negative HER2. Acridine platinum (Pt) complexes have shown specificity on the evaluated DNA sequences; 9-anilinoacridine core, which intercalates DNA, and a methyl triazene DNA-methylating moiety were also studied. Acridine thiourea gold and acridinone derivatives act against cell lines such as MDA-MB-231, SK-BR-3, and MCF-7. Benzimidazole acridine compounds demonstrated cytotoxic activity against Dual Topo and PARP-1. Quinacrine, thiazacridine, and azacridine are reported as anti-cancer agents, which have been reported in the previous decade and were addressed in this research.
- acridine
- anti-cancer
- cancer cell lines
- DNA- intercalation
- topoisomerase
- in vitro assay
1. Introduction
2. Acridine as an Anti-Tumour Agent
2.1. Acridine
The photocytotoxicity of propyl-AcrDTU against the murine leukaemia L1210 cell line has been reported. Previously, electron paramagnetic resonance (EPR) spectroscopy has been used to assess the formation of ROS by propyl-AcrDTU after irradiation. After confirming the production of ROS when UV-A light (>300 nm) was used to irradiate propyl-AcrDTU in the sight of molecular oxygen, to elucidate the mechanism of this chemical’s photocytotoxic effect, researchers focused on its intracellular location [1]. UV-Vis and fluorescence spectroscopy were used to synthesise and characterise two new tetrandrine-based receptors, as well as their bonding properties towards a range of nucleotides and ds-DNA, in water at pH = 7.2. In an intercalation-binding mode, two receptors had a strong affinity (K 105 M-1) and specificity of sequence for ds-DNA. This research included molecular modelling and single-crystal X-ray diffraction analysis. Furthermore, anti-proliferative investigations based on the derivatives of several cell lines for cancer show that the compounds have potential anti-cancer properties [2]. A promising anti-tumour drug, 9-(2′-hydroxyethylamino)-4-methyl-1-nitroacridine (C1748), was discovered to move through phase I metabolic pathways in a laboratory setting (Figure 1). The current research aimed to learn more about its metabolisation by phase II enzymes called UDP-glucuronosyltransferases (UGTs) as well as its potential for being involved in drug–drug interactions caused by UGT regulation [3].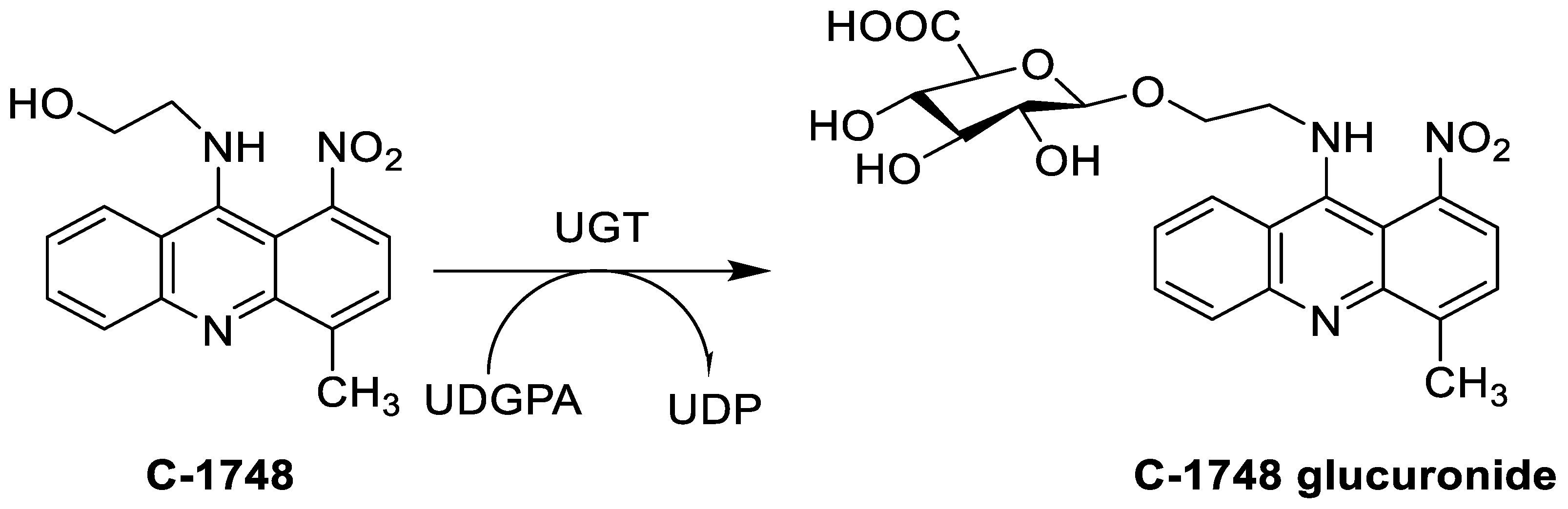
| Compound | Structure | Biological Activity | Reference | ||
|---|---|---|---|---|---|
| Propyl-AcrDTU |  |
Leukaemia L1210 cells | [1] | ||
| Tetrandrine-based receptors |  |
Anti-proliferative studies | 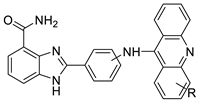 [2] [2] |
||
| Inhibitory activities against Topo and PARP-1 | [ | 9-(2′-hydroxyethylamino)-4-methyl-1-nitroacridine (C1748) | 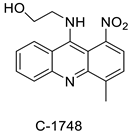 |
Phase II enzymes—UDP-glucuronosyltransferases (UGTs) | [3] |
| - |  |
Human lung adenocarcinoma cells | [4] | ||
| Acridine chalcone 1C ((2 E)-3-(acridin-9-yl)-1-(2,6-dimethoxyphenyl)prop-2-en-1-one) |  |
Human colorectal HCT116 cells | [5] | ||
| Novel spiro-acridine (E)-50-oxo-10-((3,4,5-trimethoxybenzylidene)amino)-10,50-dihydro-10H-spiro[acridine-9,20-pyrrole]40-carbonitrile (AMTAC-17) | 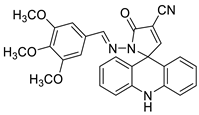 |
Anti-tumour activity of AMTAC-17 | [6] | ||
| 2-((6-Chloro-2-methoxy-acridin-9-yl)amino)-5,6,7,8-tetrahydro-4H-cyclohepta[b]-thiophene-3-carbonitrile (ACS03) | 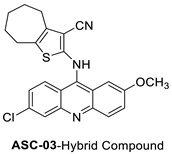 | ||||
 | |||||
| Anti-proliferative inhibitors | |||||
| [ | |||||
| 34 | |||||
| ] | |||||
2.2. 9-Amino Acridine
Four NSCLC cell lines were used to test the anti-proliferative effect of a range of acridine-based catalytic inhibitors of TOPOII (H460, A549, H2009, and H2030) [35]. The current findings in the treatment of breast cancer showed that CK0403 was more potent and effective than CK0402 against estrogen receptor-negative and HER2-overexpressing breast cancer cell lines, implying that it could be used as a breast cancer chemotherapy in the future [36]. Interactions between specific distinct moieties of 9-amino acridines and DNA were investigated and demonstrated to be important in determining the overall stabilities of DNA G-quadruplex complexes. Both 9-amino acridines were found to produce varying levels of structural stability through intercalation, although having equal binding affinities to the G-quadruplex. This distinctive trait of modifying structural stability is most likely a factor in influencing telomerase function and, as a result, the reported anti-cancer activity varied between the two 9-amino acridines [37]. The anti-malarial activity of 9-aminoacridine and artemisinin–acridine hybrid compounds against both the chloroquine sensitive but also gametocytocidal strain (NF54) and the chloroquine resistant (Dd2) Plasmodium falciparum strains was determined in vitro. CHO cell cytotoxicity, HepG2 and SH-SY5Y apoptosis, and anti-cancer efficacy against HeLa cell lines were all tested in vitro (Figure 3) [38]. 9-aminoacridine (9AA) showed specific toxicity for infectious leukemic cells regardless of their p53 status due to p53 reactivation and NF-B inhibition. It was also shown that 9AA stimulates caspase-3/7, which results in PARP cleavage. The effectiveness of 9AA in the MET-1 ATL model was also studied [39].
| Compound | Structure | Biological Activity | Reference |
|---|---|---|---|
| Acridine-based catalytic inhibitors | 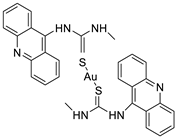 |
Anti-proliferative activity | [35] |
| Compound | Structure | Biological Activity | Reference | ||||
|---|---|---|---|---|---|---|---|
| 2-methyl-9 substituted (AS 0–8) acridines | 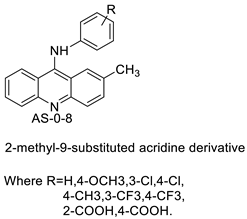 |
Lung carcinoma, breast cancer. |
[44] | ||||
| CK0403 | 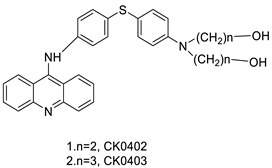 |
Treatment of breast cancer | [36] | ||||
| CK0403 | 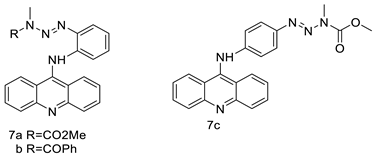 |
Anti-cancer activity | [45] | 9-amino acridines |  |
Anti-cancer activity | [37] |
| New series of 9-anilinoacridines containing phenyl-urea moieties | 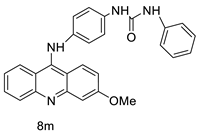 |
Novel dual Src and MEK inhibitors. | [46] | 9-aminoacridine and artemisinin–acridine |  |
Anti-malarial activity, anti-cancer activity | [38] |
| BO-1051 | 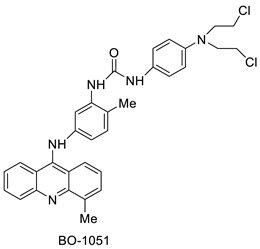 |
Against oral cancer | [47] | 9-aminoacridine (9AA) | 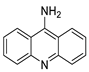 |
Anti-leukaemic cells | |
| Novel isoxazole substituted 9 –anilinoacridines | 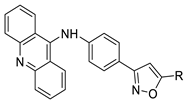 |
HER2 inhibitors. | [48] |
2.4. Acridine Thiourea Gold
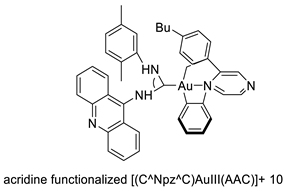
Two novel 1-acridin-9-yl-3-methylthiourea Au(I) DNA intercalators have been developed: [Au (ACRTU)2]Cl (2) and [Au(ACRTU)(PPh3)]PF6. Both complexes were extremely active in the cisplatin-sensitive A2780 human ovarian cancer cell line, with IC50 values in the sub-micromolar range. MDA-MB-231 (triple negative), SK-BR-3 (HER2+, ERα-, and ERβ-), and MCF-7 (ER+) are all cytotoxic to different phenotypes of breast cancer cell lines [49]. The synthesis of seven new cyclometalated Au(III) complexes, five of which contain an acridine moiety linked via (NO) or (NN) chelates, acyclic amino carbenes (AAC), and N-heterocyclic carbenes (NHC), was reported [50]. The anti-proliferative properties of the various complexes were investigated in vitro on a panel of cancer cells, including leukaemia, lung, and breast cancer cells. In some of the series representative substances, researchers observed a relationship between cytotoxicity and intracellular gold uptake. Some of the acridine-decorated compounds were shown to interact with ds-DNA using FRET-melting techniques (Table 4) [50].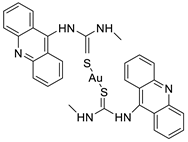
2.5. Acridine-Thiazolidinone
The interactions of three novel acridine–thiazolidinone compounds (2a–2c) using calf thymus DNA and a variety of cell lines (leukaemic cells HL-60 and L1210, and human epithelial ovarian cancer cell lines A2780) were investigated. Compounds 2a–2c had a high affinity for calf thymus DNA, with regard to binding constants ranging from 1.37 ×10−6 to 5.89 × 106 M_1 as determined by spectrofluorimetry. After 72 h of incubation, all of the investigated compounds showed substantial cytotoxic activity in vitro, with IC50 values of 1.3 ± 0.2 M (HL-60), 3.1 ± 0.4 M (L1210), and 7.7 ± 0.5 M (A2780). Acridine compounds were rapidly accumulated by cancer cells, and alterations in glutathione levels were confirmed. Cell proliferation was suppressed by the chemicals and resulted in cell-cycle arrest and cell death. Their ability to have an effect on cells was connected to thiol reactivity and DNA-binding activity [51]. N-alkylation and the Michael reaction were used to create a series of unique hybrid 5-acridin-9-ylmethylene-3-benzyl-thiazolidine-2,4-diones. A 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay was employed to assess cell viability, and DNA interaction experiments were carried out using electrochemical methods (Table 5) [52].| Compound | Structure | Biological Activity | Reference | ||
|---|---|---|---|---|---|
| Three new acridine–thiazolidinone derivatives | 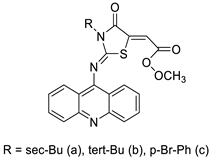 |
Leukemic cells, human epithelial ovarian cancer cell lines. | [51] | ||
| Series of novel hybrid 5-acridin-9-ylmethylene-3-benzyl-thiazolidine-2,4-diones | 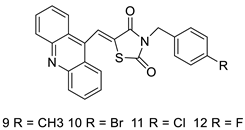 |
DNA interaction assays were performed | [52] | 57] | |
| 9-aminoacridine carboxamide | |||||
 |
DNA-targeted intercalator | [ | 40] | ||
| Anti-tumour activity | [ | 7 | ] | ||
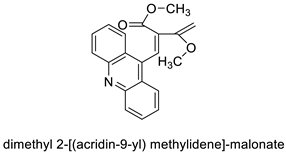
| Dimethyl 2-[(acridin-9-yl) methylidene]-malonate (LPSF/IP-81) | |||||
| Novel benzimidazole acridine derivatives | 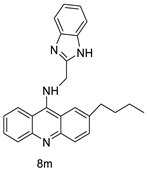 |
 |
Invasive ductal carcinoma of human breast. | [8] | |
| New DNA-targeted compounds | [ | Bis (acridine-9-carboxylate)-nitro-europium (III) dihydrate | 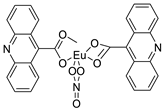 |
Anti-angiogenic and apoptotic activity against an animal model of carcinogenesis | [9] |
| 9-amino-1-nitroacridine | 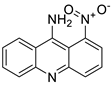 |
Pancreatic cancer | [10] | ||
| 9-amino acridine derivatives | 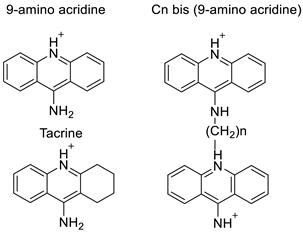 |
On alpha-adrenergic receptors | [11] | ||
| AT11-L0 |  |
Anti-proliferative activity | [12] | ||
| Novel acridine-based N-acyl-homoserine lactone (AHL) analogs |  |
2.6. Acridinone
A class of acridinones derived from the structure of podophyllo toxin were discovered as a result of a lead discovery effort aimed at less structurally complex synthetic chemicals. Wound-healing experiments using the metastatic and triple-negative breast cancer cell line MDA-MB-231 were used to test the drugs in vitro. Four compounds were discovered with IC50 values ranging from 0.294 to 1.7 μM [53]. The cytotoxic activity of a new series of 9 (10H)-acridinone-1,2,3 triazole derivatives against human breast cancer cell lines was developed, synthesised, and assessed, 2-methoxy-10-((1-(4-methoxybenzyl). The most potent compound against MCF7 cells was 1H-1,2,3-triazol-4-yl)methyl)acridin-9(10H)-one 8c (IC50 = 11.0 ± 4.8 µM), which was more potent than toposide (IC50 = 12.4 ± 4.7 µM) [54]. The designed UGT1A10’s capacity and selectivity in the glucuronidation of acridinone anticancer drugs in a cellular setting was tested. These results imply that extrahepatic UGT1A10 is involved in the metabolism and bioactivation of C-1305, and they provide a foundation for more mechanistic research into the drug’s mode of action. A translational study into the involvement of this enzyme in the regulation of C-1305 toxicity in cancer was also conducted (Table 6) [55].
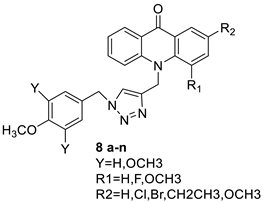
2.8. Benzoacridine
Potential anti-cancer drugs and tubulin polymerisation inhibitors were designed and synthesised. The anti-cancer activity of the synthesised compounds was assessed using the 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide (MTT) assay against eight cancer cell lines, including MCF7, A2780, HeLa, HepG2, DU145, A549, PC3, and LNCAP cancer cells, as well as normal human umbilical vein endothelial cells (HUVEC), with lapachone. With IC50 values ranging from 5.23 to 24.32 μM, some of the compounds (4c and 4g) displayed substantial cytotoxic effects on cancer cells [59]. Novel 7-substituted-5, 6-dihydrobenzo[c]acridine derivatives were designed and synthesised. According to recent biophysical research, the substitutes may effectively attach to and stabilise the c-KIT G-quadruplex with significant selectivity towards duplex DNA. The biological examination of compound 2b revealed that it might induce apoptosis by activating the caspase-3 cascade pathway [60]. A rapid one-pot microwave-assisted synthesis of novel deadly phenanthrene fused-tetra hydrodibenzo-acridinones was reported [61]. This protocol provides a broad substrate range, catalyst-free synthesis, atom-economy, simple recrystallisation, good yields, and the use of ethanol as a green solvent. The in vitro cytotoxicity of these novel compounds was tested against cervical (HeLa), prostate (PC-3), fibrosarcoma (HT-1080), and ovarian (SKOV-3) cancer cells, and they were found to be safer than the normal (Hek-293T) kidney cell line [61]. The development of a 12-arylbenzoacridine library using an approach centered on diversity resulted in non-toxic estrogenic and anti-estrogenic compounds. The estrogen receptors (ER alpha) and ER beta (IC50 lM) have a strong binding affinity for derivatives with a hydroxy group at the molecular edge, but binding to the estrogen-related receptor c (ERRc), an orphan nuclear receptor on which estrogens frequently trigger unfavourable events, was not observed. According to the findings, 12-arylbenzoacridines can be used as a new platform for the creation of selective estrogen-receptor modulators (SERMs) (Table 8) [62].| Compound | Structure | Biological Activity | Reference |
|---|---|---|---|
| Tubulin polymerisation inhibitors | 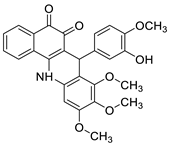 |
Eight cancer cell lines including MCF-7, A2780, HeLa, HepG2, DU145, A549, PC3, and LNCAP | [59] |
| 9-aminoacridine derivatives | |||
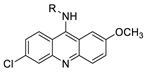 | |||
| Anti-proliferative activity | [ | 41 | ] |
| Human oral squamous carcinoma | |||
| Four acridine Pt complexes | 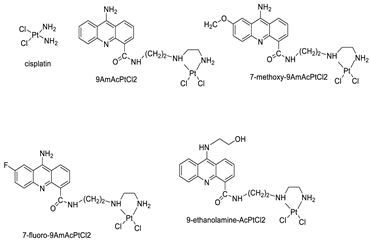 |
Detailed DNA sequence specificity | [42] |
| Small library of substituted 9-aminoacridine derivatives | 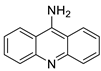 |
Ability to inhibit proliferation and induce cellular death in SCLC | [43] |
| [ | |||
| 13 | ] |
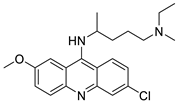
2.3. 9-Anilino Acridines
Nucleophilic substitution of 2-methyl-9-chloroacridine (AS) with aromatic amines yielded numerous 2-methyl-9 substituted (AS 0–8) acridines. The MTT assay was used to test three substances for anti-proliferative activity against A-549 (human small-cell lung carcinoma) and MCF-7 (human breast cancer) cell lines. The cytotoxicity of acridines against cancer cells was shown to be more active in the A-549 cell line than in the MCF-7 cell line [44]. The synthesis of molecular hybrids with high efficiency with a 9-anilinoacridine (9-AnA) core that intercalates DNA and a methyl triazene DNA-methylating moiety has been reported [45]. The anti-tumour activity of the chimeras was tested. In anti-proliferative experiments with multiple cancer cell lines, Chimera 7b showed the best anti-cancer activity at low micromolar IC50 values [45]. Researchers have developed and synthesised a new series of 9-anilinoacridines incorporating phenyl-urea moieties as possible new dual Src and MEK inhibitors. In vitro anti-proliferative studies on K562 and HepG-2 tumour cells revealed that the majority of the compounds were cytotoxic. According to their findings, the acridine scaffold, notably compound 8m, could be helpful in the creation of novel multi-target Src and MEK kinase inhibitors [46]. BO-1051 decreased cell viability in oral cancer cells with a low IC50, but not in normal gingival fibroblasts. BO-1051-induced tumour suppression was followed by cell-cycle arrest and downregulation of stemness genes according to cell cycle analyses. It was shown that BO-1051 had cytotoxic activity by causing the induction of autophagy and cell-cycle arrest. It was proposed that combining BO-1051 with radiation could be a viable option for oral cancer in the future [47]. In silico designs were used to create several novel isoxazole substituted 9–anilinoacridines (1a–z) with HER2 inhibitory activity. Using Schrodinger suit 2016-2, docking studies of compounds 1a–z were conducted on HER2 (PDB id-3PP0). This work adds to the evidence that isoxazole substituted 9-aminoacridine compounds could be used as HER2 inhibitors (| Against human breast cancer cell lines. | ||
| [ | 54 | ] |
| New 7-substituted-5, 6-dihydrobenzo[c]acridine derivatives |
| Compound | Structure | Biological Activity | Reference |
|---|---|---|---|
| Compound | Structure | Biological Activity | Reference |
|---|---|---|---|
| C-1748, 9-(20-hydroxyethylamino)-4-methyl-1-nitroacridine |
| Compound | Structure | Biological Activity | Biological ActivityReference |
|---|---|---|---|
 |
Anti-tumour activity | [ | 71 |
| Reference | |||
| ] | |||
| Three new thiazacridine derivatives | 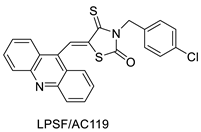 |
Induced neoplastic cell death primarily by apoptosis | |
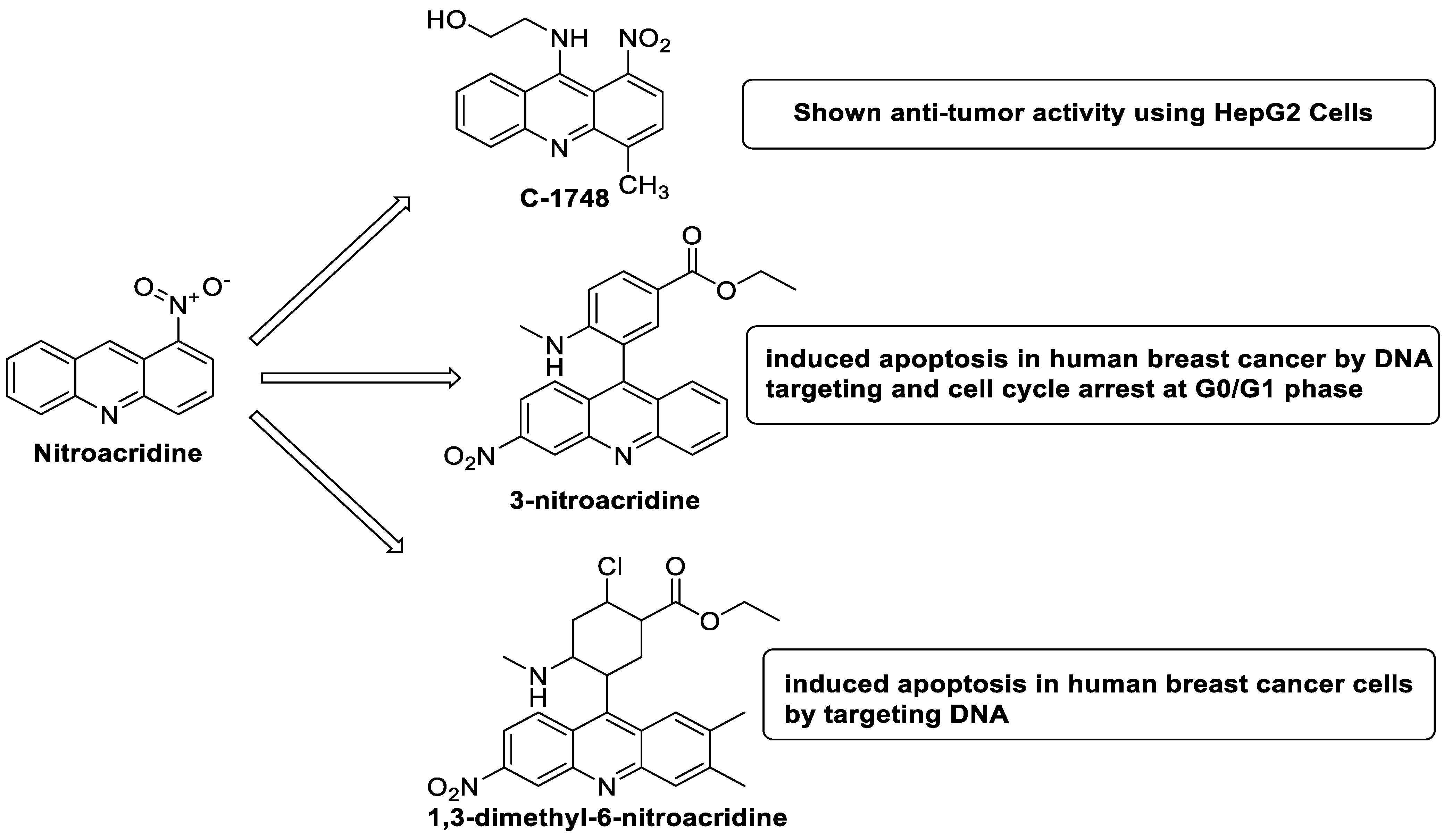
| 3-nitroacridine derivatives | 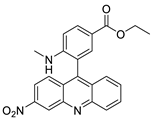 |

| Compound | Structure | Biological Activity | Reference |
|---|
| Potent Compound | Cancer Cell Lines | Control | IC50 (µM) | Reference | |
|---|---|---|---|---|---|
| Quinacrine | 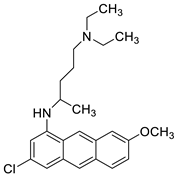 |
Human leukaemia K562cells | [77] | ||
| [ | 86 | ] | [93] | DNA-binding affinity and good inhibitory effect of tumour cell proliferation | [72] |
| C-1305 C-1311 | |||||
| New series of 1,3-dimethyl-6-nitroacridine derivatives |  |
Anti-cancer activity against four cancer cells | [73] |
2.11. Oxazine
A series of 9-anilinoacridines substituted with oxazine derivatives were synthesised and tested in vitro for antioxidant and anti-cancer activity against Daltons Lymphoma Ascites (DLA) cell growth. These compounds were found to have significant antioxidant and anti-cancer activity (inhibition of DLA cell proliferation). Compounds 5a, 5h, 5i, and 5j were the most cytotoxic, with CTC50 values ranging from 140 to 250 mg/mL. Using the Schrodinger Maestro 9.2 version, the docking studies of the synthesised derivatives were performed towards the key Topoisomerase II (1QZR). The oxazine-substituted 9-anilinoacridine derivatives 5a, 5h, 5i, and 5j have been reported to have significant anti-cancer activity as topoisomerase II inhibitors [74]. Docking investigations against topoisomerase II were conducted using new oxazine-substituted 9-anilinoacridines. Compounds 1c, 1f, and 1g had a high Glide rating. In addition, in silico ADMET screening was carried out [75]. Series of oxazine-substituted 9-anilinoacridines were synthesized, characterized, and tested for anti-cancer efficacy against Dalton’s lymphoma ascites cells utilising in vitro and in vivo methodologies. On Dalton’s lymphoma ascites cells, these conjugates showed strong antitumour activity according to the findings. Compounds 4b, 4c, 4e, and 4j were the most cytotoxic, with CTC50 values ranging from 96.5 to 190 g/mL (0.125 to 0.352 µM). The PHASE module of the Schrodinger suite was used to conduct 3D QSAR research (Table 11 and Figure 8) [76].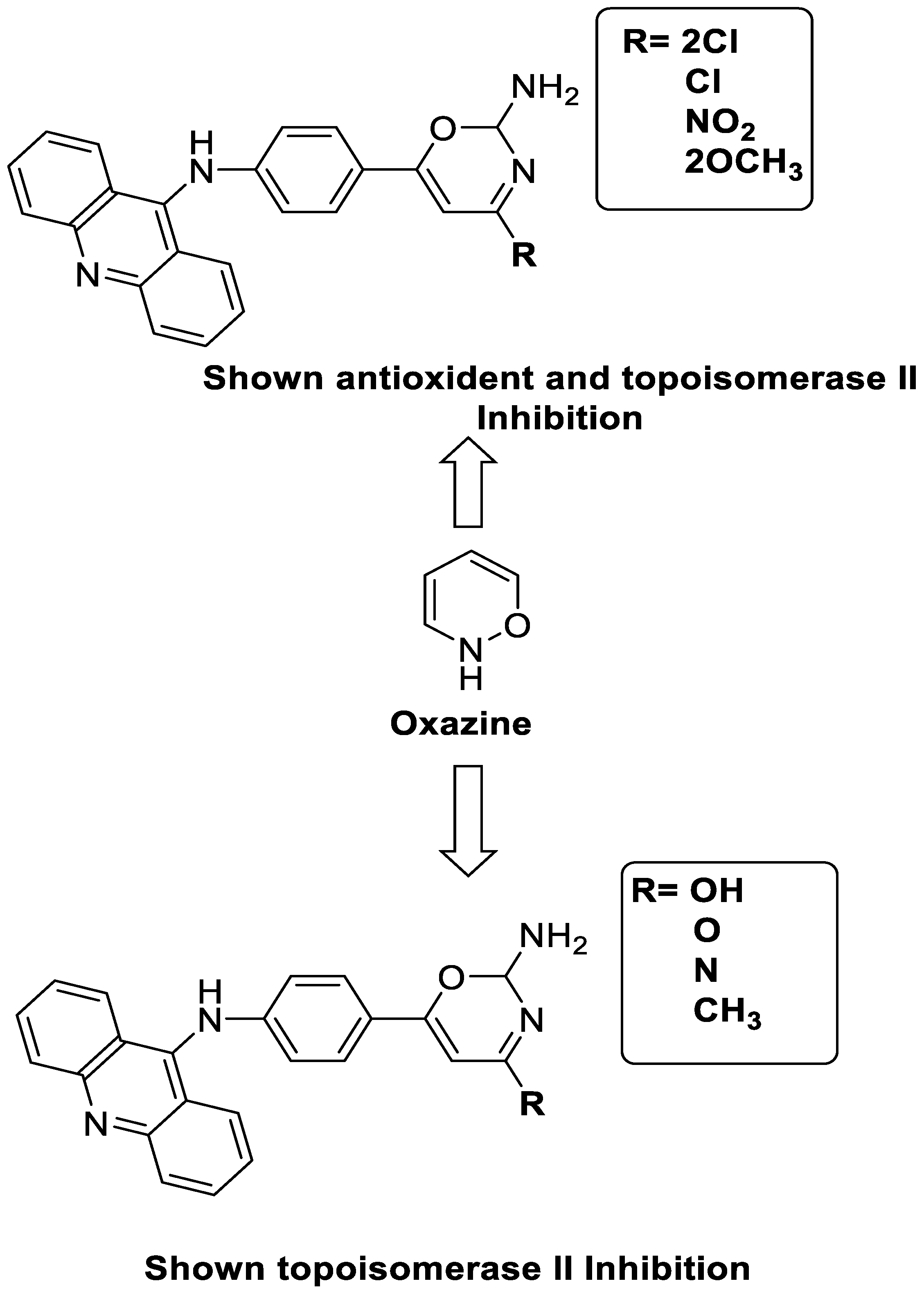
| Compound | Structure | Biological Activity | Reference | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Imidazoacridinone C-1311 | 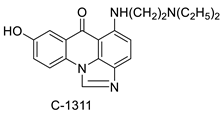 |
Anti-cancer activity | [63] | ||||||
 |
Induce apoptosis through activation of the caspase-3 cascade pathway | [ | 60] | ||||||
| A novel photo activation-based pharmacological Trojan horse approach | 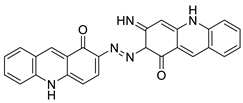 |
A novel pharmacophore comprising a DNA-targeted platinum–acridine hybrid agent | 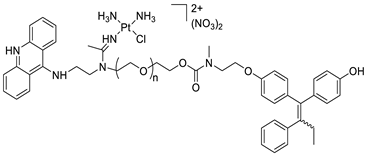 MDR tumour cells MDR tumour cells |
Breast cancer cell lines | [78] | ||||
| A new series of azaacridine derivatives | 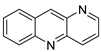 |
Potent EGFR and Src dual inhibitors | [97] | [64] | 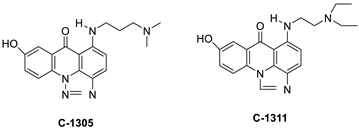 |
Phenanthrene fused-tetra hydrodibenzo-acridinones.Anti-tumour agents |  [ [ |
Cervical (HeLa), prostate (PC-3), fibrosarcoma (HT-1080), ovarian (SKOV-3) cancer cells55 | [61 |
| HaCat | |||||||||
| “ | |||||||||
| 62.18 ± 1.15 | |||||||||
| New series of acridine hydroxamic acid derivatives |  |
Topo II inhibition activity | [14] | ||||||
| 9-chloro-2-(3-(dimethylamino) propyl) pyrrolo [2,3,4-kl] acridin-1(2H)-one (LS-1-10) | 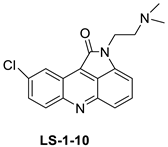 |
Colon cancer cells | [15] | ||||||
| ] | |||||||||
| 12-arylbenzoacridines | 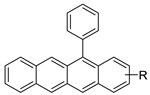 |
||||||||
| PBMC | “ | 115.2 ± 5.82 | Two iodinated acridine derivatives |  |
Targeted radionuclide therapy | [16] | |||
| Selective estrogen-receptor modulators (SERMs). | [ | ||||||||
| Cisplatin | MCF-7 | - | 12.7 ± 2.2 | [9] | [125I] ICF01035 and [125I] ICF01040 |  |
Melanoma-targeted in mice bearing B16F0 | [17] | |
| 9-Acridine carboxylic acid | “ | - | 21.5 ± 3.1 | 9-phenyl acridine (ACPH) | |||||
| Eu(III)-complex | 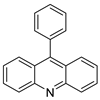 |
Anti-cancer agent | [18] | ||||||
| “ | - | 14.3 ± 2.6 | |||||||
| C-1748 | MiaPaCa-2 | - | 0.015 | [10] | Tetra hydro acridine derivatives with iodobenzoic moiety | ||||
 |
Human lung adenocarcinoma, human colorectal adenocarcinoma. | [19] | |||||||
| AsPC-1 | - | 0.075 | A series of 9-(2-(1-arylethylidene) hydrazinyl) acridine |  |
Anti-cancer activity | [20] | |||
| 9-phenylacridine (ACPH) | 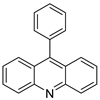 |
Antitumour activity | [21] | ||||||
| A class of acridine derivatives |  |
In vivo MM tumour growth | [22] | ||||||
| N0-(2-chloro-6-methoxy-acridin-9-yl)-2-cyano-3-(4-dimethylaminophenyl)-acrilohidrazida (ACS-AZ10). ACS-AZ10 |  |
Anti-tumour activity | [23] | ||||||
| Acridine yellow G |  |
Inhibits both EGFR and PKCs | [24] | ||||||
| 62 | ] |
2.9. Imidazoacridinone
C-1311 is an anti-tumour imidazoacridinone that inhibits DNA-reactive topoisomerase II and the FLT3 receptor tyrosine kinase. Researchers showed that C-1311 inhibits new targets such as hypoxia-inducible factor-1α (hIF1), vascular-endothelial growth factor (VeGF), and angiogenesis in this research. C-1311 is a potent inhibitor of hIF-1 α, VeGF, and angiogenesis, according to the findings, which offer fresh insights into the mechanism underlying its anti-cancer activity [63]. Photo-rupture of IA-loaded lysosomes and tumour cell lysis via production of reactive oxygen species were used in a novel photo activation-based pharmacological Trojan horse method to target and destroy MDR cancer cells. It was discovered that MDR cells’ Achilles heel is lysosomal sequestration of IAs, which may be exploited to destroy MDR tumour cells by lysosomal photo death [64]. In hepatoma cells, the effect of CYP3A4 overexpression on the cellular response was generated by the anti-tumour drug C-1311. The effect of CYP3A4 overexpression on C-1311 metabolism and the modification of CYP3A4 activity by C-1311 were also investigated. It was stated that when evaluating the possible therapeutic effects of C-1311, inter-patient variability in CYP3A4 levels should be taken into account (Figure 6) [65].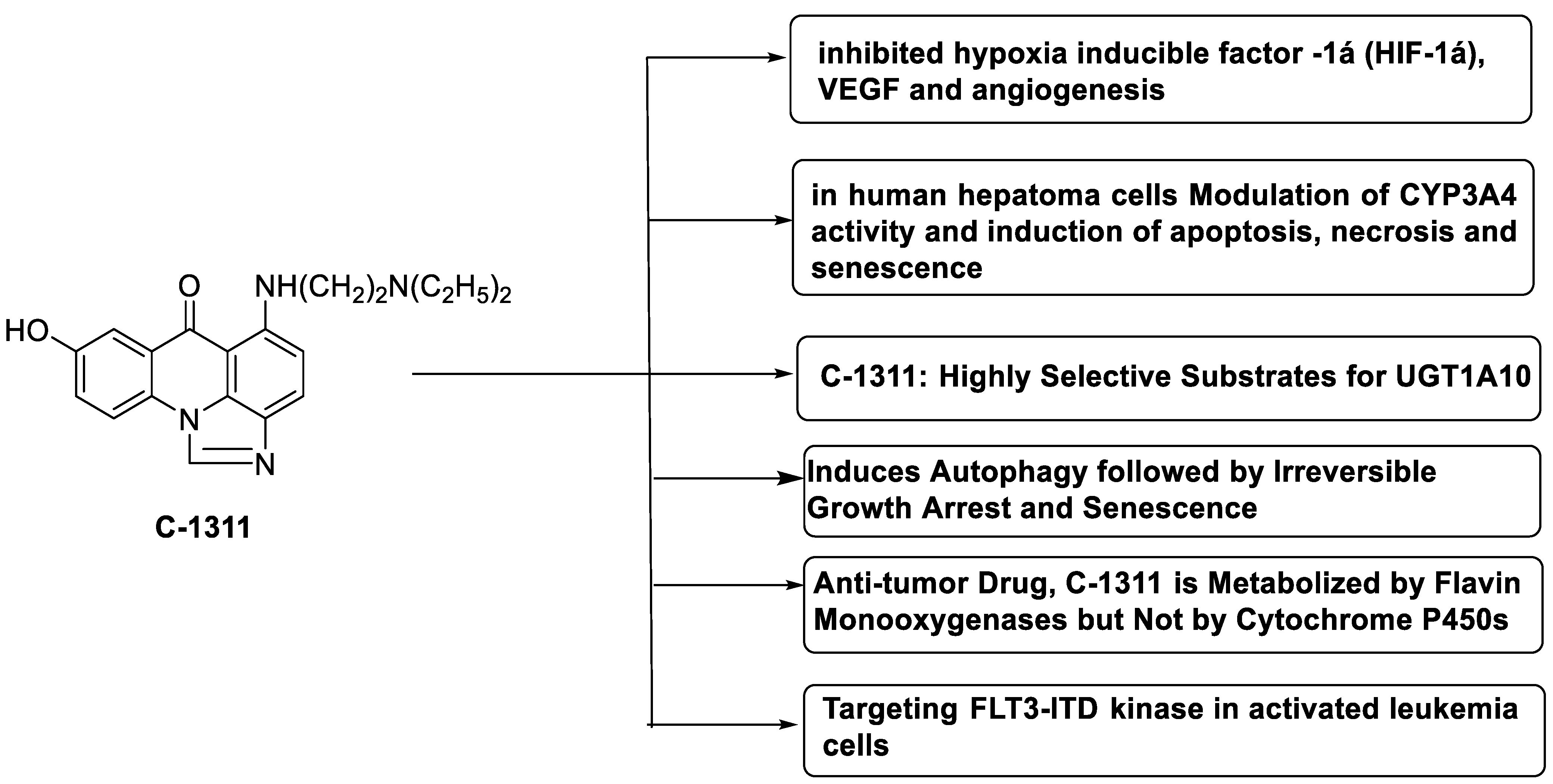
| A series of 9-anilinoacridines substituted with oxazine derivatives | 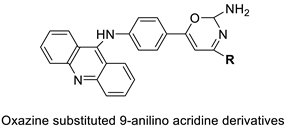 |
Antioxidant and anti-cancer activity | [74] | ||||||||
| Quinacrine | 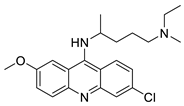 |
Human leukaemia U937 cells. | [87 | ||||||||
| Novel oxazine substituted 9-anilinoacridines | 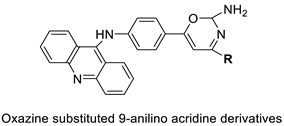 |
Topoisomerase II | [] | P3A1 | 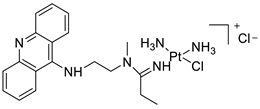 |
Cancer cells using MWCNTs as a drug carrier | [79] | ||||
| curcumin (Cur) and quinacrine (QC) | |||||||||||
| Propyl-AcrDTU | HL-60 | - | |||||||||
| An acridine and thiazolidine nucleus | 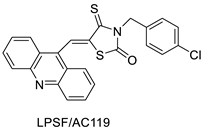 |
Tested in human colon carcinoma HCT-8 cells | [94] | ||||||||
 |
|||||||||||
| B1 | |||||||||||
| SAS Cells | |||||||||||
| - | |||||||||||
| 1.5 | |||||||||||
| [ | |||||||||||
| 13 | |||||||||||
| ] | |||||||||||
| B2 | |||||||||||
| “ | |||||||||||
| - | 11.7 | ||||||||||
| Unsymmetrical bisacridine derivatives (UAs) | |||||||||||
 |
Cytotoxicity screening against several tumour cell lines | [ | 25] | ||||||||
| 9-aryl-hexahydro-acridine-1,8-diones | |||||||||||
| 7.2–8 | [ | 1 | ] | A series of new aza-acridine analogues |  |
Anti-tumour agent | C-1311 | 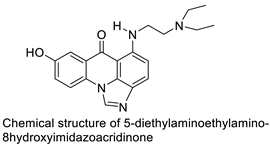 |
Anti-tumour agent C-1311in hepatoma cells | [65] | |
| B3 | “ | - | 5.3 | ||||||||
| B4 | 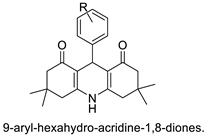 |
Anti-cancer activity against HepG2 and MCF-7 cell lines | [26] | ||||||||
| ] | Series of acridine-based catalytic inhibitors |  |
Pancreatic cancer and hTopoII catalytic inhibitors | [27] | |||||||
| 75 | ] | Four hybrid acridine-HSP90 |  |
Telomerase inhibitor BRACO-19 | [28] | ||||||
| Three new diphenyl substituted spiro triazolidine- and thiazolidinone-acridines | 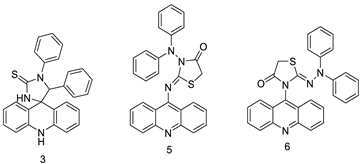 |
Interaction with calf thymus DNA | [29] | ||||||||
| Amino acid appended acridines |  |
Anti-proliferative activity, arrested cells in G0/G1 phase of the cell cycle |
[30] | ||||||||
| A novel series of tri-substituted acridines |  |
Mimicking the effects of BRACO19 | [31] | ||||||||
| DNA–polymer hybrids |  |
DNA–polymerisation | [32] | ||||||||
| A novel DNA-cleaving agent CuGGHK-Acr | 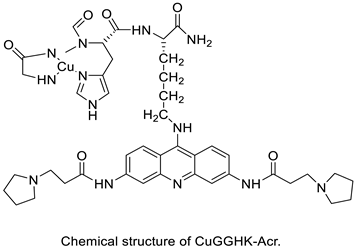 |
Targets G4 telomeric DNA | [33] | ||||||||
| A series of 9-benzyl acridine derivatives |
2.7. Benzimidazole Substituted Acridines
In human colon cancer cell lines, a novel benzimidazole acridine derivative (8m) demonstrated cytotoxic action. This derivative, 8m, activated both the intrinsic and extrinsic death pathways in a time- and concentration-dependent way according to the study. Further research into the ROS-JNK1 pathway’s mode of action revealed that it was critical in initiating 8m-induced apoptosis. Researchers also showed that 8m can upregulate DR5, which is characteristic for potent anticancer drugs [56]. Dual Topo and PARP-1 inhibitors, a series of 4-amidobenzimidazole acridines, were designed and synthesised. Compound 11l had powerful inhibiting effects on Topo and PARP-1 as well as a considerable inhibitory effect on cancer cell proliferation. According to their findings, single drugs that inhibit Topo and PARP simultaneously could be used as an alternative to cancer treatment, and 11l could be a possible lead chemical for anticancer drug discovery [57]. A series of new DNA-targeted agents (Figure 5), unique benzimidazole derivatives of acridine, were designed and synthesised (Table 7) [58].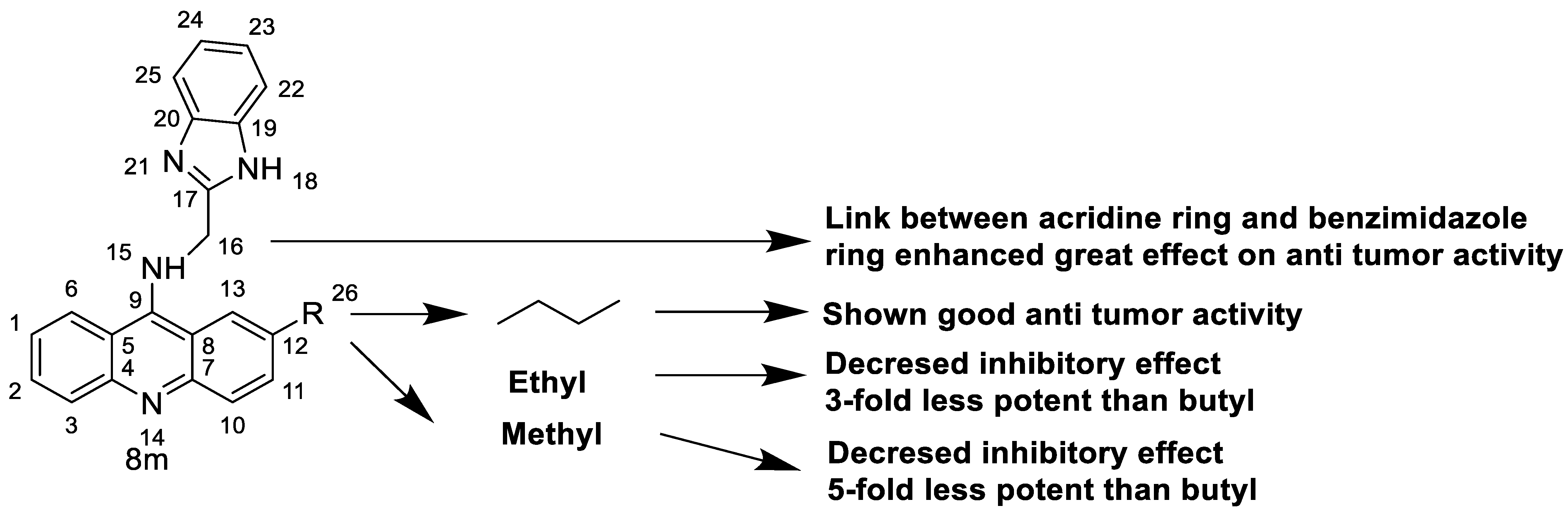
| Compound | Structure | Biological Activity | Reference |
|---|---|---|---|
| New benzimidazole acridine derivative | 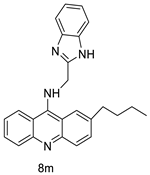 |
Human colon cancer cell lines | [56] |
| A series of 4-amidobenzimidazole acridines | |||
| A series of oxazine substituted 9-anilinoacridines | 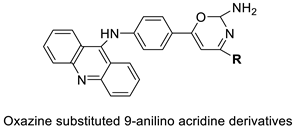 |
Anti-tumour activity on Dalton’s lymphoma ascites cells | [76] |
| Crystal structures of NQO2 containing two of the imidazoacridin-6-ones |  |
Inhibits the enzymatic function of NQO2 in cells. | [66] |
| Imidazoacridinone and triazoloacridinone |  |
Liver cancer | [67] |
| C-1311 |  |
Human non-small-cell lung cancer (NSCLC) cell lines, A549 and H460. In A549 cells | [68] |
| C-1311 and C-1330 |  |
Human live microsomal enzymes | [69] |
| Imidazoacridinone C-1311 |  |
Against FLT3-ITD kinase | [70] |
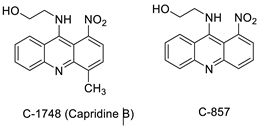
2.10. Nitroacridine
The differential in toxicity between C-1748, 9-(20-hydroxyethylamino)-4-methyl-1-nitroacridine, and its 4-demethyl counterpart, C-857, was shown to be due to changes in metabolic routes for the two chemicals; the effect of decreasing and/or hypoxic circumstances on metabolism was also examined. Finally, the importance of hypoxic circumstances and POR’s direct involvement in the metabolism of both substances was established. C-1748’s low reactivity and the stability of its metabolites, compared with C-857, are thought to have a substantial role in the compound’s reduced toxicity in animals [71]. DNA damage is caused by interactions with DNA and interference with regulatory mechanisms. The structure–activity connection revealed that substituents on position 9 of 3-nitroacridine derivatives were crucial for DNA-binding and anti-proliferative effects. Compounds2.12. Platinum-Acridine Anti-Cancer Agents
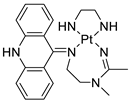
A validated genome-wide screening approach was used to examine the spectrum of activity of a library of non-classical platinum-acridine hybrid substances in Saccharomyces cerevisiae and the distantly related yeast Schizo saccharomyces pombe. Chemo-genomic profiles of S. cerevisiae and S. pombe revealed that many platinum-acridines cause DNA damage that differs from cisplatin even though they require different DNA-repair modules [77]. Carbamate-coupling chemistry was used to synthesise a new pharmacophore containing a DNA-targeted platinum–acridine hybrid agent and estrogen receptor-targeted 4-hydroxy-N-desmethyl tamoxifen (endoxifen) and perform its analysis in breast cancer cell lines [78]. MWCNTs coated with a non-classical platinum chemotherapeutic agent ([PtCl(NH3)2(L)] Cl (P3A1; L = N-(2-(acridin-9-ylamino)ethyl)-N-methylproprionimidamide) and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[amino(sn-glycero-3-phosphoethanolamine-N-[amino(sn-glycero-3-phosphoethanolamine-N-[amino(sn-g (DSPE-mPEG) were reported. This finding suggests that employing MWCNTs as a medication carrier to deliver P3A1 to cancer cells could be advantageous for cancer chemotherapy and photothermal therapy [79]. The configuration of unique seven-membered, sterically overloaded chelates [Pt(en)(L/L0)](NO3)2 (4a/4b) from effective hybrid anti-tumour compounds [PtCl(en)(LH/L0H)](NO3)2 (3a/3b), where en is ethane-1,2-diamine and L(H) and L0(H) are (protonated) N-(2-(Compounds 3a and 3b have IC50 values of 12 (2 and 2.8(0.3 nM, respectively)))), and inhibit H460 lung cancer cell proliferation [80]. Carboxylic acid ester-group-containing platinum–acridine hybrid agents (Cpd: 2.12.5) were created. In the cell lines for ovarian cancer (OVCAR-3) and breast cancer (MCF-7, MDA-MB231), the most effective derivatives and parent chemicals that had not been modified were demonstrated as having up to 6-fold greater action than cisplatin. Cell proliferation was inhibited 80- and 150-fold at nanomolar doses in pancreatic (PANC1) and non-small-cell lung (NSCLC, NCI-H460) cancer cells, respectively [80]. A platinum–acridine hybrid agent [PtCl(en)(L)](NO3)2 (complex 1, en = ethane-1,2-diamine, L = 1-[2-(acridin-9-ylamino)ethyl]-1,3-dimethylthiourea was studied using a combination of biophysical, biochemical, and computational techniques to determine mechanistic distinctions between it and a much more effective second-generation analogue. N-methylpropionamidine is a specific type of N-methylpropionamidine [81]. Five NSCLC cell lines were resynthesised and assessed, indicating big cell, squamous cell, and adenocarcinomas. 7-Aminobenz[c] acridine was identified as a promising scaffold in a hybrid drug (P1–B1) and showed 32-fold enhanced tolerability in mice compared with the parent platinum–acridine (P1–A1) while maintaining sub-micromolar activity in numerous DNA-repair-proficient and p53-mutant cancer models [82]. Binds generated by the platinum–acridine agent [Pt Cl(en)(N-(2-(acridin-9-ylamino)ethyl)-N-methylpropionimidamide)] were structurally characterised using high-performance liquid chromatography (HPLC) in combination with electrospray mass spectrometry (LC-ESMS). In cell-free DNA, (NO3)2 (compound 1) was present [83]. [PtCl(en)(LH)], a platinum-acridine anti-cancer agent, [en = ethane-1,2-diamine, LH = N-(2-(acridin-9-ylamino)ethyl)-N-methylpropion imidamide], an acridinium cation, and (NO3)2 (1) [en = ethane-1,2-diamine, LH = N-(2-(acridin-9-ylamino)ethyl)-N-methylpropion imidamide], acridinium, were studied. The cytotoxic potency and cell-kill mechanisms of and the therapeutic medication cisplatin were investigated in chemo-resistant cell lines such as non-small-cell lung cancer (NSCLC). Compound 1 revealed a 40200-fold cytotoxic increase compared with cisplatin in the three studied cell lines (NCI-H460, NCI-H522, and NCI-H1435) at inhibitory doses approaching the low-nano molar range (Table 12 and Figure 9) [84].
| Compound | Structure | Biological Activity | Reference | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Non-classical platinum-acridine hybrid agents |  |
Distinct modules of DNA repair | |||||||
| [ | 97 | ] | |||||||
| MAnT | HeLa cervical cancer cell line | Tetrandrine | 2.74 | Induced breast epithelial transformed (MCF-10A-Tr) generated metastatic cells | Series of new thiazacridine agents[ |  88 88 |
HepG2 cells | [95] | |
| [ | 2 | ] | |||||||
| C-1748 | HT29 Cell | Sorafenib | 39.7 | [3] | [Pt(en)(L/L0)](NO3)2 [PtCl(en)(LH/L0H)](NO3)2 |
 |
Inhibits H460 lung cancer cell proliferation | [80] | |
| ] | |||||||||
| Quinacrine | 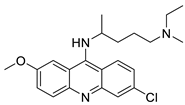 |
Thiazacridine and imidazacridine derivatives | 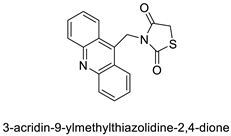 Inhibition of PARP-1PARylation Inhibition of PARP-1PARylation |
Tumours suppressors in some cancer cell lines[89] | [96] | Platinum−acridine hybrid agents | 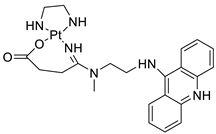 |
Anti-cancer activity | [81] |
| Quinacrine | 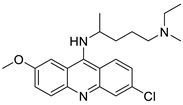 |
Cytotoxic effect of QC on DLBCL cells | [90] | Platinum acridine hybrid agent [PtCl(en)(L)](NO3)2 | 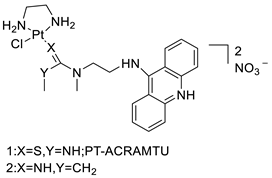 |
Delineate mechanistic differences between the platinum acridine hybrid agent | |||
| Quinacrine | 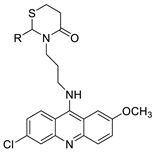 | [ | 82] | ||||||
| Against different cancer cell types | [ | 91 | 7-aminobenz[c] acridine | 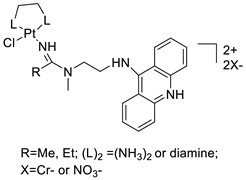 |
Tested in five NSCLC cell lines | [83] | |||
| Platinum–acridine agent | 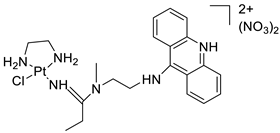 |
Structurally characterised | [84] | ||||||
| Platinum-acridine anti-cancer agent [PtCl(en)(LH)](NO3)2 | 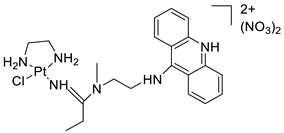 |
Chemo resistant non-small-cell lung cancer (NSCLC) cell lines | [85] |
2.13. Quinacrine
In human leukaemia K562 cells, the cytotoxic effect and mechanism of quinacrine activity were investigated. Quinacrine promoted apoptosis in K562 cells as well as ROS production, mitochondrial depolarisation, and BCL2L1 and BCL2 down-regulation. Quinacrine-induced cell death in K562 cells, according to the results, was mediated by mitochondrial changes caused by p38 MAPK-mediated BCL2 down-regulation and suppression of ERK/c-Jun-mediated BCL2L1 expression (Figure 10) [85].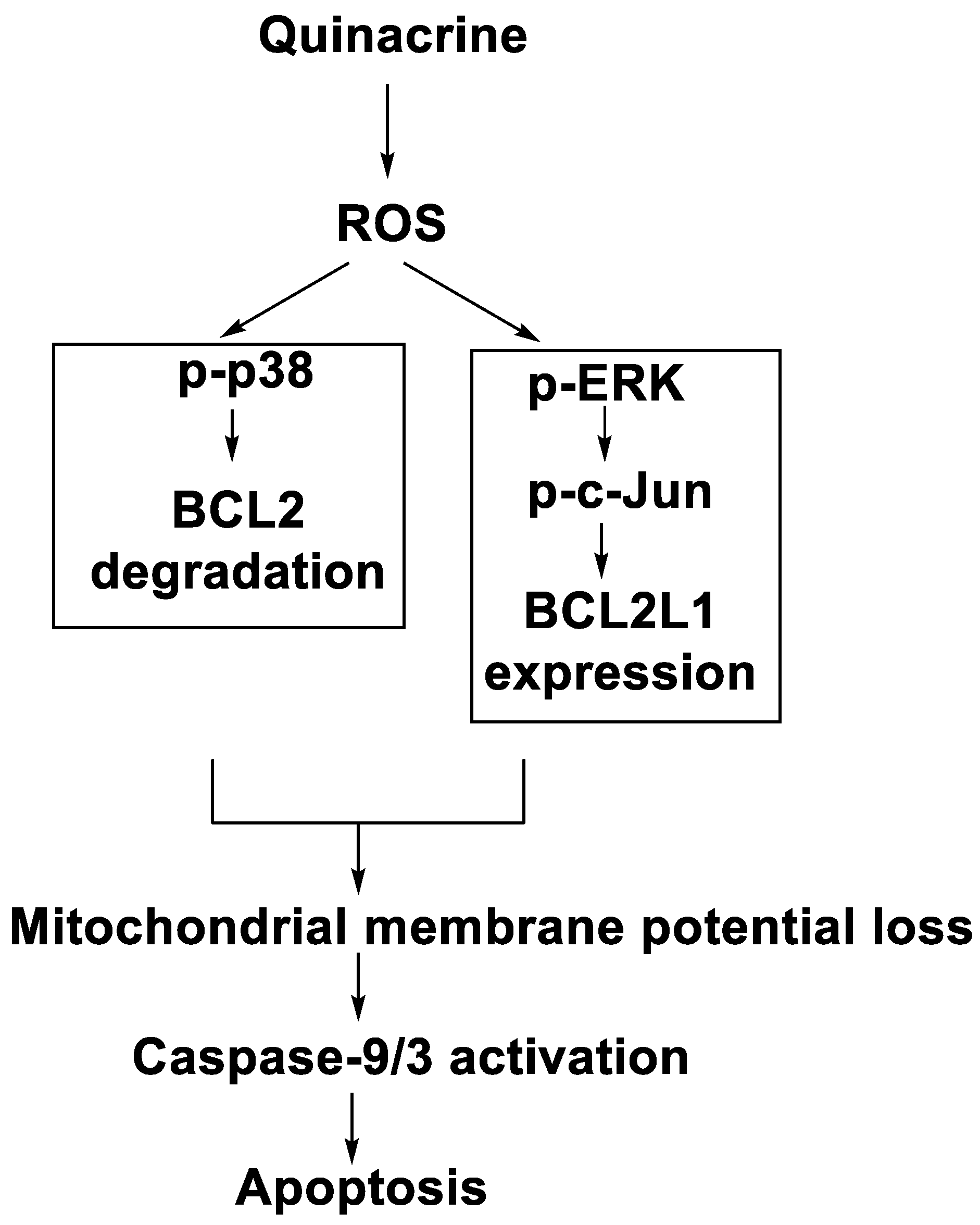
| ] |
| [ |
| 92 |
| ] |
2.14. Thiazacridine
Three novel thiazacridine compounds were synthesised and their anti-proliferative activities were evaluated. Three new thiazacridine compounds were produced and described using a three-step synthetic reaction: (Z)-5-acridin-9-ylmethylene-3-(4-methyl-benzyl)-4-thioxo-thiazolidin-2-one (LPSF/AC-99), (Z)-5-acridin-9-ylmethylene-3-(4-chloro-benzyl)-4-thioxo-thiazolidin-2-one (LPSF/AC-119), and (Z)-5-acridin-9-ylmethylene-3-. Colorimetric assays were used to test toxicity and selectivity. According to the findings, none of the chemicals were toxic to normal human cells and caused neoplastic cell death primarily through apoptosis [92]. A novel cytotoxic drug with acridine and thiazolidine nuclei the cytotoxic activity of four ATZDs was examined in human colon cancer HCT-8 cells: (5Z)-5-acridin-9-ylmethylene-3-(4-methylbenzyl)-thiazolidine-2,4-dione-AC-4; (5ZE)-5-acridin-9-ylmethylene-3-(4-methylbenzyl)-thiazolidine-2,4-dione-AC-4 -3-(4-bromobenzyl)thiazolidine-2,4-dione-AC-7;(5Z) -5-(acridin-9-ylmethylene) -3-(4-chlorobenzyl) -3-(4-chlorobenzyl)-1,3-thiazolidine-2,4-dione-AC-10; and -1,3-thiazolidine-2,4-dione-AC-10 (5ZE) AC-23 is a -5-(acridin-9-ylmethylene)-3-(4-fluoro-benzyl)-1,3-thiazolidine-2,4-dione. All of the ATZDs tested inhibited HCT-8 cell proliferation in a concentration- and time-dependent manner. According to the findings, ATZD inhibited the activity of DNA Topo isomerase I and affected tumour cell apoptosis via apoptotic pathways [93]. A number of novel thiazacridine compounds have been produced and tested as anti-cancer agents, both in terms of cytotoxicity and selectivity. All compounds had cytotoxic activity and selectivity according to the cytotoxicity assay. The most promising molecule was 3-acridin-9-ylmethyl-5-(5-bromo-1H-indol-3ylmethylene)-thiazolidine-2,4-dione (LPSF/AA29—7a), with IC50 values ranging from 0.25 to 68.03 mM depending on cell lineage. In HepG2 cells, the IC50 value for -thiazolidine-2,4-dione (LPSF/AA36—7b; 46.95 mM) was the lowest. None of the produced compounds were shown to be harmful to normal cells (IC50 > 100 mM) [94]. In several cancer cell lines, thiazacridine and imidazacridine derivatives have shown promise as tumour suppressors. That was the case in binding studies of 5-acridin-9-ylmethylidene-3-amino-2thioxo-thiazolidin-4-one, 5-acridin-9-ylmethylidene-2-thioxo-thiazolidin-4-one, 5-acridin9-ylmethylidene-2-thioxo-imidazolidin-4-one, and 3-acridin-9-ylmethylidene-2-thioxo-imid, except for 5-acridin-9-ylmethylidene-2-thioxo-1,3-thiazolidin-4-one, which showed inhibitory activity against human topoisomerase I. These findings shed light on the process by which imidazacridines and thiazacridines bind to DNA (Table 14 and Figure 11) [95].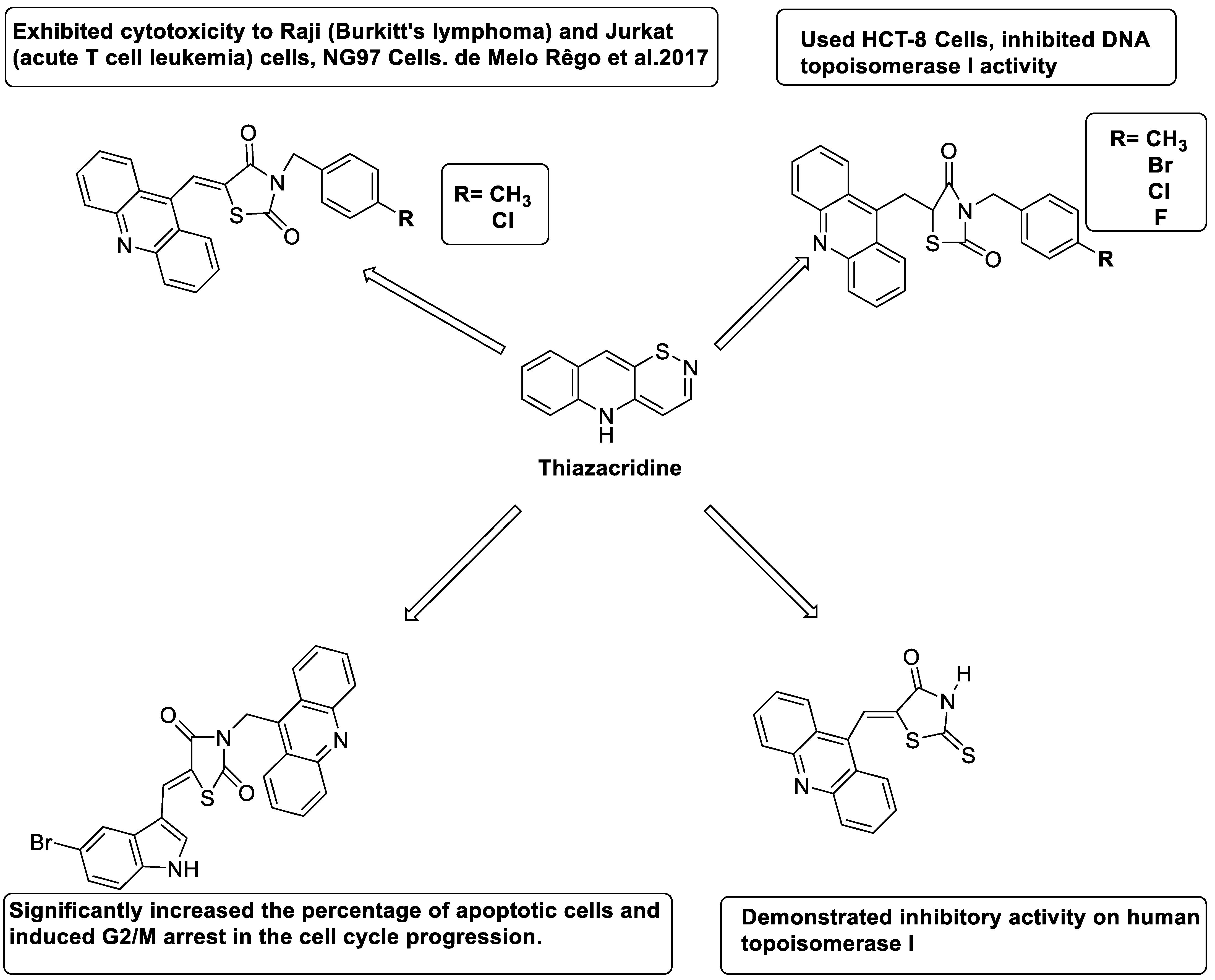
| Compound | Structure |
|---|
2.15. Azacridine
A new category of azaacridine compounds, (Figure 12), which are effective EGFR and Src dual inhibitors, was designed and synthesised. According to the findings, the majority of the azaacridine compounds synthesised had good anti-proliferative activity against K562 and A549 cells. The representative 13b inhibited EGFR and Src activity, as well as tumour-cell invasion and apoptosis [96]. A variety of novel azaacridine analogues with a basic side chain were produced and their anti-proliferative efficacy was tested. The compounds had minimal biological activity against three cancer cell lines when compared with their acridine equivalents. This was thought to be due to the compounds’ hydrolytic instability in aqueous conditions (Table 15 and Table 16) [97].| Cmpd- | ||||
| 1 | ||||
| A549 Cells | Etoposide | 15 | [ | 4] |
| Cmpd-2 | A549 Cells | “ | 10 | |
| Cmpd-3 | A549 Cells | “ | 15 | |
| Cmpd-4 | A549 Cells | “ | 10 | |
| ACSO3 | HCT-116 Cells | Doxorubicin | 23.11 ± 1.03 | [7] |
| HeLa | “ | ˃50 | ||
| MCF-7 | “ | ˃50 | ||
| K562 | “ | ˃50 | ||
| HL-60 | “ | ˃50 | ||
| “ | ||||
| - | ||||
| 18.0 | ||||
| B5 | ||||
| “ | - | 9.4 | ||
| 80c | U937 HCT-116 |
m-AMSA | 0.90 | [14] |
| LS-1-10 | DLD1 Cells | Amsacrine | 1.31 | [15] |
| ACPH | A375 | Camptothecin | 20.74 | [18] |
| 1i | A549 HT-29 |
Etoposide | 59.12–14.87 17.32–5.90 |
[19] |
| 4b,4d,4e | HeLa, HepG2, HEK 239 |
Camptothecin | 18.89–52.64 18.7–108.34 31.38–277.13 |
[20] |
| C2 | malignant glioma and other human cancers. | erlotinib | 0.75–5 | [24] |
| C-1311 | DU-145 HCT-116 MBA-MB-231 |
Gemcitabine Erlotinib |
0.01–0.03 0.01–0.03 0.01–0.03 |
[25] |
| 4b 4f 4g 4i 4j |
HepG2 MCF-7 |
Doxorubicin | 1.4 ± 0.11 4.7 ± 0.09 2.2 ± 0.09 5.3 ± 0.16 4.8 ± 0.12 4.4 ± 0.10 2.6 ± 0.11 5.9 ± 0.15 1.6 ± 0.14 5.0 ± 0.18 |
[26] |
| SR374 SR375 SR361 SR362 |
MCF7 A549 GIST48 WI38 |
- | 1.6, 0.4, 2.9, <0.1 0.3, 0.1, 0.5, 0.1 1.5, 1.6, 2.8, 2.3 7.4, 4.5, 10.3, 0.7 |
[28] |
| Cmpd 5 | HL-60 | Acridine | 25.87 32.16 |
[29] |
| Cmpd 13 Cmpd 15 Cmpd 16 Cmpd 17 Cmpd 18 |
HT-29 | BRACO19 | 13.17–12.55 41.12–27.38 69.99–22.12 48.80–23.25 44.89–33.23 |
[31] |
| CuGGHK-Acr GGHK-Acr Ho-Acr |
HuH-7 MCF-7 Caco2 |
- | 9.8 ± 2.3 26.6 ± 2.2 16.7 ± 1.6 |
[33] |
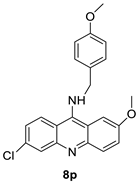
| 8p | A549 Cells | Doxorubicin Etoposide Amsacrine |
0.61 ± 0.06 | [34] |
| 9AmAcPtCl2 | HeLa Cells | Cisplatin | 0.4 | [40] |
| 7r | HepG-2 MCF-7 |
Colchicin | 4.2 23.8 |
[41] |
| 7b | H1299 WM264 HCT116 |
Cisplatin | 2.9 0.8 15.4 |
[45] |
| 8m | K562 HepG-2 |
Imatinib | 4.08 ± 0.14 9.41 ± 1.09 |
[46] |
| BO-1051 | SAS Cells OECM1 |
- | 2.39 1.97 |
[47] |
| Cmpd 2 | A2780 MDA-MB-231 SK-BR-3 MCF-7 |
CDDP AuC1PPh3 |
0.88 ± 0.20 2.75 ± 0.40 3.62 ± 0.14 5.32 ± 0.65 |
[50] |
| Complex 11 | A549 MCF-7 HL60 |
Cisplatin | 7.6 ± 0.6 1.5 ± 0.1 1.1 ± 0.1 |
[50] |
| 2C | HL-60 L1210 A2780 |
Cisplatin | 1.3 ± 0.2 3.1 ± 0.4 7.7 ± 0.5 |
[51] |
| Cmpd-9 | PC-3 COLO-205 |
Amsacrine | 5.5–9.5 8.6–42.3 |
[52] |
| Cmpd-6 Cmpd-7 Cmpd-9 Cmpd-10 |
MDA-MB-231 DU-145 |
Colchicine | 3 ± 1, 3.2 ± 0.7 0.190 ± 0.007 1.1 ± 0.2 1.0 ± 0.3 0.11 ± 0.02 0.12 ± 0.02 |
[53] |
| 8c | MCF-7 T-47D MDA-MB-231 |
etoposide | 11 ± 4.8 14.5 ± 5.2 16.6 ± 5.9 |
[54] |
| C-1305 C-1311 |
KB-3 | - | 0.263 ± 0.016 0.106 ± 0.014 |
[55] |
| 8m | SW480 HCT116 |
- | 6.77 ± 0.19 3.33 ± 0.02 |
[56] |
| 11L | PARP-1 MCF-7 |
Olaparib m-AMSA |
0.45 ± 0.03 2.14 ± 0.92 |
[57] |
| 8I | K562 HepG-2 |
Colchicine Imatinib |
2.68 8.11 |
[58] |
| 4g | HUVEC LNCAP |
Β-Lapachone | 49.19 ± 2.3 18.54 ± 2.11 |
[59] |
| 2b | HeLa K562 A549 |
- | 3.2 9.2 5.7 |
[60] |
| 8m | SKOV-3 | Cisplatin | 0.24 ± 0.05 | [61] |
| Cmpd-1 | MCF-7 | Tamoxifen citrate | 0.951 | [62] |
| 6a1 | HCT116 | NQO2 | 14 ± 4 | [66] |
| C-1311 | MV4-11 MOLM13 |
- | 0.03 ± 0.01 0.04 ± 0.02 |
[70] |
| Cmpd-2 | MCF-7 MDA-MB-231 SGC7901 MGC803 |
- | 6.79 ± 2.01 6.46 ± 2.19 17.25 ± 3.84 10.94 ± 2.26 |
[72] |
| Cmpd-1 Cmpd-6 |
MCF-7 MDA-MB-231 SGC7901 MGC803 |
- | 8.83 ± 0.98 10.02 ± 0.78 41.47 ± 3.24 23.96 ± 1.54 |
[73] |
| 5a | DLA Cells | ascorbic acid | 20.03 ± 0.2583 | [74] |
| 8 | MCF-7 MDA-MB-231 |
Tamoxifen | 1.6 ± 0.4 13.2 ± 0.1 |
[78] |
| Cmpd-3 | NCI-H460 OVCAR-3 PANC-1 |
Cisplatin | 0.008 ± 0.002 1.1 ± 0.1 0.09 ± 0.01 |
[80] |
| 3a 3b |
H460 | - | 12 ± 2 2.8 ± 0.3 |
[81] |
| P1-A1 P1-B1 P1-B2 |
NCI-H460 | - | 0.0052 ± 0.0001 0.24 ± 0.01 2.4 ± 0.5 |
[83] |
| Cmpd-1 | NCI-H460 NCI-H522 |
Cisplatin | 8 ± 2 18 ± 2 |
[85] |
| QC | OCI-Ly01 SU-DHL-8 |
Quinacrine | 1.8 2 |
[90] |
| Cmpd-25 | MDA-MB-468 MDA-MB-231 MCF-7 184B5 |
Cisplatin Quinacrine |
1.73 ± 0.80 2.80 ± 1.30 0.69 ± 0.41 4.96 ± 0.24 |
[91] |
| Cmpd-11 | MDA-MB-468 MDA-MB-231 MCF-7 184B5 |
Cisplatin Quinacrine |
2.40 ± 1.01 1.92 ± 0.20 1.24 ± 0.51 16.16 ± 0.81 |
[92] |
| LPSF/AC-119 | Raji Jurkat |
Amsacrine | 0.6 1.53 |
[93] |
| AC-4 AC-7 AC-10 AC-23 |
HCT-8 | Amsacrine | 3.1 5.3 3.6 2.3 |
[94] |
| 7a 7b |
HepG2 | Amsacrine | 68.03 48.63 |
[95] |
| 13b | K562 A549 |
Imatinib | 0.22 ± 0.03 0.253 ± 0.16 |
[97] |
References
- Cisáriková, A.; Barbieriková, Z.; Janovec, L.; Imrich, J.; Hunáková, L.; Bačová, Z.; Paulíková, H. Acridin-3, 6-dialkyldithiourea hydrochlorides as new photosensitizers for photodynamic therapy of mouse leukaemia cells. Bioorg. Med. Chem. 2016, 24, 2011–2022.
- Calvillo-Páez, V.; Sotelo-Mundo, R.R.; Leyva-Peralta, M.; Gálvez-Ruiz, J.C.; Corona-Martínez, D.; Moreno-Corral, R.; Escobar-Picos, R.; Höpfl, H.; Juárez-Sánchez, O.; Lara, K.O. Synthesis, spectroscopic, physicochemical and structural characterization of tetrandrine-based macrocycles functionalized with acridine and anthracene groups: DNA binding and anti-proliferative activity. Chem.-Biol. Interact. 2018, 286, 34–44.
- Mróz, A.; Ryska, I.; Sominko, H.; Bejrowska, A.; Mazerska, Z. Drug-drug interaction potential of antitumor acridine agent C-1748: The substrate of UDP-glucuronosyltransferases 2B7, 2B17 and the inhibitor of 1A9 and 2B7. Pharmacol. Rep. 2018, 70, 972–980.
- Szymański, P.; Olszewska, P.; Mikiciuk-Olasik, E.; Różalski, A.; Maszewska, A.; Markiewicz, L.; Cuchra, M.; Majsterek, I. Novel tetrahydroacridine and cyclopentaquinoline derivatives with fluorobenzoic acid moiety induce cell cycle arrest and apoptosis in lung cancer cells by activation of DNA damage signaling. Tumor Biol. 2017, 39, 1010428317695011.
- Takac, P.; Kello, M.; Vilkova, M.; Vaskova, J.; Michalkova, R.; Mojzisova, G.; Mojzis, J. Antiproliferative effect of acridine chalcone is mediated by induction of oxidative stress. Biomolecules 2020, 10, 345.
- Silva, D.K.F.; Duarte, S.S.; Lisboa, T.M.; Ferreira, R.C.; Lopes, A.L.d.; Carvalho, D.; Rodrigues-Mascarenhas, S.; da Silva, P.M.; Segundo, M.A.; de Moura, R.O. Antitumor effect of a novel spiro-acridine compound is associated with up-regulation of Th1-type responses and antiangiogenic action. Molecules 2020, 25, 29.
- Lisboa, T.; Silva, D.; Duarte, S.; Ferreira, R.; Andrade, C.; Lopes, A.L.; Ribeiro, J.; Farias, D.; Moura, R.; Reis, M. Toxicity and antitumor activity of a thiophene–acridine hybrid. Molecules 2020, 25, 64.
- Almeida, S.M.V.; Silva, L.P.B.G.; Lima, L.R.A.; Botelho, S.P.S.; Lima, M.d.C.; Pitta, I.d.; Beltrão, E.I.C.; Junior, L.B.C. Dimethyl-2--malonate as fluorescent probe for histochemical analysis. Microsc. Res. Tech. 2017, 80, 608–614.
- Azab, H.A.; Hussein, B.H.; El-Azab, M.F.; Gomaa, M.; El-Falouji, A.I. Bis (acridine-9-carboxylate)-nitro-europium (III) dihydrate complex a new apoptotic agent through Flk-1 down regulation, caspase-3 activation and oligonucleosomes DNA fragmentation. Bioorg. Med. Chem. 2013, 21, 223–234.
- Borowa-Mazgaj, B.; Mróz, A.; Augustin, E.; Paluszkiewicz, E.; Mazerska, Z. The overexpression of CPR and P450 3A4 in pancreatic cancer cells changes the metabolic profile and increases the cytotoxicity and pro-apoptotic activity of acridine antitumor agent, C-1748. Biochem. Pharmacol. 2017, 142, 21–38.
- Campbell, A.P.; Wakelin, L.P.; Denny, W.A.; Finch, A.M. Homobivalent conjugation increases the allosteric effect of 9-aminoacridine at the α1-adrenergic receptors. Mol. Pharmacol. 2017, 91, 135–144.
- Carvalho, J.; Lopes-Nunes, J.; Lopes, A.C.; Campello, M.P.C.; Paulo, A.; Queiroz, J.A.; Cruz, C. Aptamer-guided acridine derivatives for cervical cancer. Org. Biomol. Chem. 2019, 17, 2992–3002.
- Chai, H.; Hazawa, M.; Hosokawa, Y.; Igarashi, J.; Suga, H.; Kashiwakura, I. Novel acridine-based N-acyl-homoserine lactone analogs induce endoreduplication in the human oral squamous carcinoma cell line SAS. Biol. Pharm. Bull. 2012, 35, 1257–1263.
- Chen, J.; Li, D.; Li, W.; Yin, J.; Zhang, Y.; Yuan, Z.; Gao, C.; Liu, F.; Jiang, Y. Design, synthesis and anticancer evaluation of acridine hydroxamic acid derivatives as dual Topo and HDAC inhibitors. Bioorg. Med. Chem. 2018, 26, 3958–3966.
- Fu, W.; Li, X.; Lu, X.; Zhang, L.; Li, R.; Zhang, N.; Liu, S.; Yang, X.; Wang, Y.; Zhao, Y. A novel acridine derivative, LS-1-10 inhibits autophagic degradation and triggers apoptosis in colon cancer cells. Cell Death Dis. 2017, 8, e3086.
- Gardette, M.; Papon, J.; Bonnet, M.; Desbois, N.; Labarre, P.; Wu, T.-D.; Miot-Noirault, E.; Madelmont, J.-C.; Guerquin-Kern, J.-L.; Chezal, J.-M. Evaluation of new iodinated acridine derivatives for targeted radionuclide therapy of melanoma using 125I, an Auger electron emitter. Investig. New Drugs 2011, 29, 1253–1263.
- Gardette, M.; Viallard, C.; Paillas, S.; Guerquin-Kern, J.-L.; Papon, J.; Moins, N.; Labarre, P.; Desbois, N.; Wong-Wah-Chung, P.; Palle, S. Evaluation of two 125I-radiolabeled acridine derivatives for Auger-electron radionuclide therapy of melanoma. Investig. New Drugs 2014, 32, 587–597.
- Ghosh, R.; Bhowmik, S.; Guha, D. 9-phenyl acridine exhibits antitumour activity by inducing apoptosis in A375 cells. Mol. Cell. Biochem. 2012, 361, 55–66.
- Girek, M.; losiński, K.K.; Grobelski, B.; Pizzimenti, S.; Cucci, M.A.; Daga, M.; Barrera, G.; Pasieka, Z.; Czarnecka, K.; Szymański, P. Novel tetrahydroacridine derivatives with iodobenzoic moieties induce G0/G1 cell cycle arrest and apoptosis in A549 non-small lung cancer and HT-29 colorectal cancer cells. Mol. Cell. Biochem. 2019, 460, 123–150.
- Haider, M.R.; Ahmad, K.; Siddiqui, N.; Ali, Z.; Akhtar, M.J.; Fuloria, N.; Fuloria, S.; Ravichandran, M.; Yar, M.S. Novel 9-(2-(1-arylethylidene) hydrazinyl) acridine derivatives: Target Topoisomerase 1 and growth inhibition of HeLa cancer cells. Bioorg. Chem. 2019, 88, 102962.
- Hansda, S.; Ghosh, G.; Ghosh, R. 9-phenyl acridine photosensitizes A375 cells to UVA radiation. Heliyon 2020, 6, e04733.
- Jiang, D.; Tam, A.B.; Alagappan, M.; Hay, M.P.; Gupta, A.; Kozak, M.M.; Solow-Cordero, D.E.; Lum, P.Y.; Denko, N.C.; Giaccia, A.J. Acridine derivatives as inhibitors of the ire1α–xbp1 pathway are cytotoxic to human multiple myeloma. Mol. Cancer Ther. 2016, 15, 2055–2065.
- Mangueira, V.M.; Batista, T.M.; Brito, M.T.; de Sousa, T.K.G.; da Cruz, R.M.D.; de Abrantes, R.A.; Veras, R.C.; de Medeiros, I.A.; Medeiros, K.K.d.; Pereira, A.L.d. A new acridine derivative induces cell cycle arrest and antiangiogenic effect on Ehrlich ascites carcinoma model. Biomed. Pharmacother. 2017, 90, 253–261.
- Qi, Q.; He, K.; Yoo, M.-H.; Chan, C.-B.; Liu, X.; Zhang, Z.; Olson, J.J.; Xiao, G.; Wang, L.; Mao, H. Acridine yellow G blocks glioblastoma growth via dual inhibition of epidermal growth factor receptor and protein kinase C kinases. J. Biol. Chem. 2012, 287, 6113–6127.
- Paluszkiewicz, E.; Horowska, B.; Borowa-Mazgaj, B.; Peszyńska-Sularz, G.; Paradziej-Lukowicz, J.; Augustin, E.; Konopa, J.; Mazerska, Z. Design, synthesis and high antitumor potential of new unsymmetrical bisacridine derivatives towards human solid tumors, specifically pancreatic cancers and their unique ability to stabilize DNA G-quadruplexes. Eur. J. Med. Chem. 2020, 204, 112599.
- Ramesh, K.B.; Pasha, M.A. Study on one-pot four-component synthesis of 9-aryl-hexahydro-acridine-1, 8-diones using SiO2–I as a new heterogeneous catalyst and their anticancer activity. Bioorg. Med. Chem. Lett. 2014, 24, 3907–3913.
- Raza, A.; Jacobson, B.A.; Benoit, A.; Patel, M.R.; Jay-Dixon, J.; Hiasa, H.; Ferguson, D.M.; Kratzke, R.A. Novel acridine-based agents with topoisomerase II inhibitor activity suppress mesothelioma cell proliferation and induce apoptosis. Investig. New Drugs 2012, 30, 1443–1448.
- Roe, S.; Gunaratnam, M.; Spiteri, C.; Sharma, P.; Alharthy, R.D.; Neidle, S.; Moses, J.E. Synthesis and biological evaluation of hybrid acridine-HSP90 ligand conjugates as telomerase inhibitors. Org. Biomol. Chem. 2015, 13, 8500–8504.
- Salem, O.M.; Vilková, M.; JanoĿková, J.; Jendželovskỳ, R.; FedoroĿko, P.; Žilecká, E.; Kašpárková, J.; Brabec, V.; Imrich, J.; Kožurková, M. New spiro tria (thia) zolidine acridines as topoisomerase inhibitors, DNA binders and cytostatic compounds. Int. J. Biol. Macromol. 2016, 86, 690–700.
- Singh, P.; Kumar, A.; Sharma, A.; Kaur, G. Identification of amino acid appended acridines as potential leads to anti-cancer drugs. Bioorg. Med. Chem. Lett. 2015, 25, 3854–3858.
- Ungvarsky, J.; Plsikova, J.; Janovec, L.; Koval, J.; Mikes, J.; Mikesová, L.; Harvanova, D.; Fedorocko, P.; Kristian, P.; Kasparkova, J. Novel trisubstituted acridines as human telomeric quadruplex binding ligands. Bioorg. Chem. 2014, 57, 13–29.
- Wilks, T.R.; Pitto-Barry, A.; Kirby, N.; Stulz, E.; O’Reilly, R.K. Construction of DNA–polymer hybrids using intercalation interactions. Chem. Commun. 2014, 50, 1338–1340.
- Yu, Z.; Han, M.; Cowan, J.A. Toward the Design of a Catalytic Metallodrug: Selective Cleavage of G-Quadruplex Telomeric DNA by an Anticancer Copper–Acridine–ATCUN Complex. Angew. Chem. 2015, 127, 1921–1925.
- Zhang, W.; Zhang, B.; Yang, T.; Wang, N.; Gao, C.; Tan, C.; Liu, H.; Jiang, Y. Synthesis and antiproliferative activity of 9-benzylamino-6-chloro-2-methoxy-acridine derivatives as potent DNA-binding ligands and topoisomerase II inhibitors. Eur. J. Med. Chem. 2016, 116, 59–70.
- Ferguson, D.M.; Jacobson, B.A.; Jay-Dixon, J.; Patel, M.R.; Kratzke, R.A.; Raza, A. Targeting topoisomerase II activity in NSCLC with 9-aminoacridine derivatives. Anticancer Res. 2015, 35, 5211–5217.
- Sun, Y.-W.; Chen, K.-Y.; Kwon, C.-H.; Chen, K.-M. CK0403, a 9-aminoacridine, is a potent anti-cancer agent in human breast cancer cells. Mol. Med. Rep. 2016, 13, 933–938.
- Aguilera, R.F.; Artali, R.; Benoit, A.; Gómez, R.G.; Casadellà, R.E.i.; Ferguson, D.M.; Sham, Y.Y.; Mazzini, S. Structure and stability of human telomeric G-quadruplex with preclinical 9-amino acridines. PLoS ONE 2013, 8, E57701.
- Joubert, J.P.; Smit, F.J.; Plessis, L.D.; Smith, P.J.; N’Da, D.D. Synthesis and in vitro biological evaluation of aminoacridines and artemisinin–acridine hybrids. Eur. J. Pharm. Sci. 2014, 56, 16–27.
- Ju, W.; Zhang, M.; Petrus, M.; Maeda, M.; Pise-Masison, C.A.; Waldmann, T.A. Combination of 9-aminoacridine with Campath-1H provides effective therapy for a murine model of adult T-cell leukaemia. Retrovirology 2014, 11, 43.
- Kava, H.W.; Murray, V. CpG methylation increases the DNA binding of 9-aminoacridine carboxamide Pt analogues. Bioorg. Med. Chem. 2016, 24, 4701–4710.
- Luan, X.; Gao, C.; Zhang, N.; Chen, Y.; Sun, Q.; Tan, C.; Liu, H.; Jin, Y.; Jiang, Y. Exploration of acridine scaffold as a potentially interesting scaffold for discovering novel multi-target VEGFR-2 and Src kinase inhibitors. Bioorganic Med. Chem. 2011, 19, 3312–3319.
- Paul, M.; Murray, V. The sequence selectivity of DNA-targeted 9-aminoacridine cisplatin analogues in a telomere-containing DNA sequence. JBIC J. Biol. Inorg. Chem. 2011, 16, 735–743.
- Ryan, E.; Blake, A.J.; Benoit, A.; David, M.F.; Robert, A.K. Efficacy of substituted 9-aminoacridine derivatives in small cell lung cancer. Investig. New Drugs 2013, 31, 285–292.
- Kumar, R.; Sharma, A.; Sharma, S.; Silakari, O.; Singh, M.; Kaur, M. Synthesis, characterization and antitumor activity of 2-methyl-9-substituted acridines. Arab. J. Chem. 2017, 10, S956–S963.
- Walunj, D.; Egarmina, K.; Tuchinsky, H.; Shpilberg, O.; Hershkovitz-Rokah, O.; Grynszpan, F.; Gellerman, G. Expedient synthesis and anticancer evaluation of dual-action 9-anilinoacridine methyl triazene chimeras. Chem. Biol. Drug Des. 2021, 97, 237–252.
- Cui, Z.; Li, X.; Li, L.; Zhang, B.; Gao, C.; Chen, Y.; Tan, C.; Liu, H.; Xie, W.; Yang, T. Design, synthesis and evaluation of acridine derivatives as multi-target Src and MEK kinase inhibitors for anti-tumor treatment. Bioorg. Med. Chem. 2016, 24, 261–269.
- Lo, W.-L.; Chu, P.-Y.; Lee, T.-H.; Su, T.-L.; Chien, Y.; Chen, Y.-W.; Huang, P.-I.; Tseng, L.-M.; Tu, P.-H.; Kao, S.-Y. A Combined DNA-affinic molecule and N-mustard alkylating agent has an anti-cancer effect and induces autophagy in oral cancer cells. Int. J. Mol. Sci. 2012, 13, 3277–3290.
- Kalirajan, R.; Pandiselvi, A.; Gowramma, B.; Balachandran, P. In-silico design, ADMET screening, MM-GBSA binding free energy of some novel isoxazole substituted 9-anilinoacridines as HER2 inhibitors targeting breast cancer. Curr. Drug Res. Rev. Former. Curr. Drug Abus. Rev. 2018, 11, 118–128.
- Perez, S.A.; de Haro, C.; Vicente, C.; Donaire, A.; Zamora, A.; Zajac, J.; Kostrhunova, H.; Brabec, V.; Bautista, D.; Ruiz, J. New acridine thiourea gold (I) anticancer agents: Targeting the nucleus and inhibiting vasculogenic mimicry. ACS Chem. Biol. 2017, 12, 1524–1537.
- Williams, M.R.; Bertrand, B.; Fernandez-Cestau, J.; Waller, Z.A.; O’Connell, M.A.; Searcey, M.; Bochmann, M. Acridine-decorated cyclometallated gold (III) complexes: Synthesis and anti-tumour investigations. Dalton Trans. 2018, 47, 13523–13534.
- Paulíková, H.; Vantová, Z.; Hunáková, L.; Čižeková, L.; Čarná, M.; Kožurková, M.; Sabolová, D.; Kristian, P.; Hamul’aková, S.; Imrich, J. DNA binding acridine–thiazolidinone agents affecting intracellular glutathione. Bioorg. Med. Chem. 2012, 20, 7139–7148.
- Barros, F.W.; Silva, T.G.; Pitta, M.G.d.; Bezerra, D.P.; Costa-Lotufo, L.V.; de Moraes, M.O.; Pessoa, C.; de Moura, M.A.F.; de Abreu, F.C.; de Lima, M.d.C.A. Synthesis and cytotoxic activity of new acridine-thiazolidine derivatives. Bioorg. Med. Chem. 2012, 20, 3533–3539.
- Magalhaes, L.G.; Marques, F.B.; da Fonseca, M.B.; Rogerio, K.R.; Graebin, C.S.; Andricopulo, A.D. Discovery of a series of acridinones as mechanism-based tubulin assembly inhibitors with anticancer activity. PLoS ONE 2016, 11, e0160842.
- Mohammadi-Khanaposhtani, M.; Safavi, M.; Sabourian, R.; Mahdavi, M.; Pordeli, M.; Saeedi, M.; Ardestani, S.K.; Foroumadi, A.; Shafiee, A.; Akbarzadeh, T. Design, synthesis, in vitro cytotoxic activity evaluation, and apoptosis-induction study of new 9 (10H)-acridinone-1, 2, 3-triazoles. Mol. Divers. 2015, 19, 787–795.
- Pawlowska, M.; Chu, R.; Fedejko-Kap, B.; Augustin, E.; Mazerska, Z.; Radominska-Pandya, A.; Chambers, T.C. Metabolic transformation of antitumor acridinone C-1305 but not C-1311 via selective cellular expression of UGT1A10 increases cytotoxic response: Implications for clinical use. Drug Metab. Dispos. 2013, 41, 414–421.
- Chen, K.; Chu, B.; Liu, F.; Li, B.; Gao, C.; Li, L.; Sun, Q.; Shen, Z.; Jiang, Y. New benzimidazole acridine derivative induces human colon cancer cell apoptosis in vitro via the ROS-JNK signaling pathway. Acta Pharmacol. Sin. 2015, 36, 1074–1084.
- Yuan, Z.; Chen, S.; Chen, C.; Chen, J.; Chen, C.; Dai, Q.; Gao, C.; Jiang, Y. Design, synthesis and biological evaluation of 4-amidobenzimidazole acridine derivatives as dual PARP and Topo inhibitors for cancer therapy. Eur. J. Med. Chem. 2017, 138, 1135–1146.
- Gao, C.; Li, B.; Zhang, B.; Sun, Q.; Li, L.; Li, X.; Chen, C.; Tan, C.; Liu, H.; Jiang, Y. Synthesis and biological evaluation of benzimidazole acridine derivatives as potential DNA-binding and apoptosis-inducing agents. Bioorg. Med. Chem. 2015, 23, 1800–1807.
- Behbahani, F.S.; Tabeshpour, J.; Mirzaei, S.; Golmakaniyoon, S.; Tayarani-Najaran, Z.; Ghasemi, A.; Ghodsi, R. Synthesis and biological evaluation of novel benzo acridine-diones as potential anticancer agents and tubulin polymerization inhibitors. Arch. Der Pharm. 2019, 352, 1800307.
- Guo, Q.-L.; Su, H.-F.; Wang, N.; Liao, S.-R.; Lu, Y.-T.; Ou, T.-M.; Tan, J.-H.; Li, D.; Huang, Z.-S. Synthesis and evaluation of 7-substituted-5, 6-dihydrobenzo acridine derivatives as new c-KIT promoter G-quadruplex binding ligands. Eur. J. Med. Chem. 2017, 130, 458–471.
- Kumar, N.P.; Sharma, P.; Reddy, T.S.; Shankaraiah, N.; Bhargava, S.K.; Kamal, A. Microwave-assisted one-pot synthesis of new phenanthrene fused-tetrahydrodibenzo-acridinones as potential cytotoxic and apoptosis inducing agents. Eur. J. Med. Chem. 2018, 151, 173–185.
- Torikai, K.; Koga, R.; Liu, X.; Umehara, K.; Kitano, T.; Watanabe, K.; Oishi, T.; Noguchi, H.; Shimohigashi, Y. Design and synthesis of benzoacridines as estrogenic and anti-estrogenic agents. Bioorg. Med. Chem. 2017, 25, 5216–5237.
- Paradziej-Lukowicz, J.; Skwarska, A.; Peszyńska-Sularz, G.; Brillowska-Dąbrowska, A.; Konopa, J. Anticancer imidazoacridinone C-1311 inhibits hypoxia-inducible factor-1α (HIF-1α), vascular endothelial growth factor (VEGF) and angiogenesis. Cancer Biol. Ther. 2011, 12, 586–597.
- Adar, Y.; Stark, M.; Bram, E.E.; Nowak-Sliwinska, P.; van den Bergh, H.; Szewczyk, G.; Sarna, T.; Skladanowski, A.; Griffioen, A.W.; Assaraf, Y.G. Imidazoacridinone-dependent lysosomal photodestruction: A pharmacological Trojan horse approach to eradicate multidrug-resistant cancers. Cell Death Dis. 2012, 3, e293.
- Augustin, E.; Paw\lowska, M.; Polewska, J.; Potega, A.; Mazerska, Z. Modulation of CYP3A4 activity and induction of apoptosis, necrosis and senescence by the anti-tumour imidazoacridinone C-1311 in human hepatoma cells. Cell Biol. Int. 2013, 37, 109–120.
- Dunstan, M.S.; Barnes, J.; Humphries, M.; Whitehead, R.C.; Bryce, R.A.; Leys, D.; Stratford, I.J.; Nolan, K.A. Novel inhibitors of NRH: Quinone oxidoreductase 2 (NQO2): Crystal structures, biochemical activity, and intracellular effects of imidazoacridin-6-ones. J. Med. Chem. 2011, 54, 6597–6611.
- Fedejko-Kap, B.; Bratton, S.M.; Finel, M.; Radominska-Pandya, A.; Mazerska, Z. Role of human UDP-glucuronosyltransferases in the biotransformation of the triazoloacridinone and imidazoacridinone antitumor agents C-1305 and C-1311: Highly selective substrates for UGT1A10. Drug Metab. Dispos. 2012, 40, 1736–1743.
- Polewska, J.; Skwarska, A.; Augustin, E.; Konopa, J. DNA-damaging imidazoacridinone C-1311 induces autophagy followed by irreversible growth arrest and senescence in human lung cancer cells. J. Pharmacol. Exp. Ther. 2013, 346, 393–405.
- Potega, A.; Dabrowska, E.; Niemira, M.; Kot-Wasik, A.; Ronseaux, S.; Henderson, C.J.; Wolf, C.R.; Mazerska, Z. The imidazoacridinone antitumor drug, C-1311, is metabolized by flavin monooxygenases but not by cytochrome P450s. Drug Metab. Dispos. 2011, 39, 1423–1432.
- Skwarska, A.; Augustin, E.; Beffinger, M.; Wojtczyk, A.; Konicz, S.; Laskowska, K.; Polewska, J. Targeting of FLT3-ITD kinase contributes to high selectivity of imidazoacridinone C-1311 against FLT3-activated leukemia cells. Biochem. Pharmacol. 2015, 95, 238–252.
- Wiśniewska, A.; Niemira, M.; Jagiełło, K.; Potęga, A.; Świst, M.; Henderson, C.; Skwarska, A.; Augustin, E.; Konopa, J.; Mazerska, Z. Diminished toxicity of C-1748, 4-methyl-9-hydroxyethylamino-1-nitroacridine, compared with its demethyl analog, C-857, corresponds to its resistance to metabolism in HepG2 cells. Biochem. Pharmacol. 2012, 84, 30–42.
- Zhou, Q.; You, C.; Zheng, C.; Gu, Y.; Gu, H.; Zhang, R.; Wu, H.; Sun, B. 3-Nitroacridine derivatives arrest cell cycle at G0/G1 phase and induce apoptosis in human breast cancer cells may act as DNA-target anticancer agents. Life Sci. 2018, 206, 1–9.
- Zhou, Q.; Wu, H.; You, C.; Gao, Z.; Sun, K.; Wang, M.; Chen, F.; Sun, B. 1,3-dimethyl-6-nitroacridine derivatives induce apoptosis in human breast cancer cells by targeting DNA. Drug Dev. Ind. Pharm. 2019, 45, 212–221.
- Kalirajan, R.; Kulshrestha, V.; Sankar, S.; Jubie, S. Docking studies, synthesis, characterization of some novel oxazine substituted 9-anilinoacridine derivatives and evaluation for their antioxidant and anticancer activities as topoisomerase II inhibitors. Eur. J. Med. Chem. 2012, 56, 217–224.
- Kalirajan, R.; Sankar, S.; Jubie, S.; Gowramma, B. Molecular docking studies and in-silico ADMET screening of some novel oxazine substituted 9-anilinoacridines as topoisomerase II inhibitors. Indian J. Pharm. Educ. Res. 2017, 51, 110–115.
- Kalirajan, R.; Kulshrestha, V.; Sankar, S. Synthesis, characterization and antitumour activity of some novel oxazine substituted 9-anilinoacridines and their 3D-QSAR studies. Indian J. Pharm. Sci. 2018, 80, 921–929.
- Cheung-Ong, K.; Song, K.T.; Ma, Z.; Shabtai, D.; Lee, A.Y.; Gallo, D.; Heisler, L.E.; Brown, G.W.; Bierbach, U.; Giaever, G. Comparative chemogenomics to examine the mechanism of action of DNA-targeted platinum-acridine anticancer agents. ACS Chem. Biol. 2012, 7, 1892–1901.
- Ding, S.; Qiao, X.; Kucera, G.L.; Bierbach, U. Design of a platinum–acridine–endoxifen conjugate targeted at hormone-dependent breast cancer. Chem. Commun. 2013, 49, 2415–2417.
- Fahrenholtz, C.D.; Ding, S.; Bernish, B.W.; Wright, M.L.; Zheng, Y.; Yang, M.; Yao, X.; Donati, G.L.; Gross, M.D.; Bierbach, U. Design and cellular studies of a carbon nanotube-based delivery system for a hybrid platinum-acridine anticancer agent. J. Inorg. Biochem. 2016, 165, 170–180.
- Graham, L.A.; Suryadi, J.; West, T.K.; Kucera, G.L.; Bierbach, U. Synthesis, aqueous reactivity, and biological evaluation of carboxylic acid ester-functionalized platinum–acridine hybrid anticancer agents. J. Med. Chem. 2012, 55, 7817–7827.
- Kostrhunova, H.; Malina, J.; Pickard, A.J.; Stepankova, J.; Vojtiskova, M.; Kasparkova, J.; Muchova, T.; Rohlfing, M.L.; Bierbach, U.; Brabec, V. Replacement of a thiourea with an amidine group in a monofunctional platinum–acridine antitumor agent. Effect on DNA interactions, DNA adduct recognition and repair. Mol. Pharm. 2011, 8, 1941–1954.
- Pickard, A.J.; Liu, F.; Bartenstein, T.F.; Haines, L.G.; Levine, K.E.; Kucera, G.L.; Bierbach, U. Redesigning the DNA-Targeted Chromophore in Platinum–Acridine Anticancer Agents: A Structure–Activity Relationship Study. Chem.-Eur. J. 2014, 20, 16174–16187.
- Qiao, X.; Zeitany, A.E.; Wright, M.W.; Essader, A.S.; Levine, K.E.; Kucera, G.L.; Bierbach, U. Analysis of the DNA damage produced by a platinum–acridine antitumor agent and its effects in NCI-H460 lung cancer cells. Metallomics 2012, 4, 645–652.
- Smyre, C.L.; Saluta, G.; Kute, T.E.; Kucera, G.L.; Bierbach, U. Inhibition of DNA synthesis by a platinum–acridine hybrid agent leads to potent cell kill in nonsmall cell lung cancer. ACS Med. Chem. Lett. 2011, 2, 870–874.
- Changchien, J.-J.; Chen, Y.-J.; Huang, C.-H.; Cheng, T.-L.; Lin, S.-R.; Chang, L.-S. Quinacrine induces apoptosis in human leukaemia K562 cells via p38 MAPK-elicited BCL2 down-regulation and suppression of ERK/c-Jun-mediated BCL2L1 expression. Toxicol. Appl. Pharmacol. 2015, 284, 33–41.
- Huang, C.-H.; Lee, Y.-C.; Chen, Y.-J.; Wang, L.-J.; Shi, Y.-J.; Chang, L.-S. Quinacrine induces the apoptosis of human leukaemia U937 cells through FOXP3/miR-183/β-TrCP/SP1 axis-mediated BAX upregulation. Toxicol. Appl. Pharmacol. 2017, 334, 35–46.
- Nayak, D.; Tripathi, N.; Kathuria, D.; Siddharth, S.; Nayak, A.; Bharatam, P.V.; Kundu, C. Quinacrine and curcumin synergistically increased the breast cancer stem cells death by inhibiting ABCG2 and modulating DNA damage repair pathway. Int. J. Biochem. Cell Biol. 2020, 119, 105682.
- Siddharth, S.; Nayak, D.; Nayak, A.; Das, S.; Kundu, C.N. ABT-888 and quinacrine induced apoptosis in metastatic breast cancer stem cells by inhibiting base excision repair via adenomatous polyposis coli. DNA Repair 2016, 45, 44–55.
- Yang, S.; Sheng, L.; Xu, K.; Wang, Y.; Zhu, H.; Zhang, P.; Mu, Q.; Ouyang, G. Anticancer effect of quinacrine on diffuse large B-cell lymphoma via inhibition of MSI2-NUMB signaling pathway Corrigendum in/10.3892/mmr. 2020.11813. Mol. Med. Rep. 2018, 17, 522–530.
- Solomon, V.R.; Pundir, S.; Le, H.-T.; Lee, H. Design and synthesis of novel quinacrine--thiazinan-4-one hybrids for their anti-breast cancer activity. Eur. J. Med. Chem. 2017, 143, 1028–1038.
- Solomon, V.R.; Almnayan, D.; Lee, H. Design, synthesis and characterization of novel quinacrine analogs that preferentially kill cancer over non-cancer cells through the down-regulation of Bcl-2 and up-regulation of Bax and Bad. Eur. J. Med. Chem. 2017, 137, 156–166.
- Rego, M.J.d.; de Moura, R.O.; Jacob, I.T.; Lins, T.U.L.e.; Pereira, M.C.; Lima, M.d.C.A.; Galdino-Pitta, M.R.; Pitta, I.d.; Pitta, M.G.d. Synthesis and anticancer evaluation of thiazacridine derivatives reveals new selective molecules to hematopoietic neoplastic cells. Comb. Chem. High Throughput Screen. 2017, 20, 713–718.
- Barros, F.W.; Bezerra, D.P.; Ferreira, P.M.; Cavalcanti, B.C.; Silva, T.G.; Pitta, M.G.; de Lima, M.d.C.; Galdino, S.L.; Pitta, I.d.; Costa-Lotufo, L.V. Inhibition of DNA topoisomerase I activity and induction of apoptosis by thiazacridine derivatives. Toxicol. Appl. Pharmacol. 2013, 268, 37–46.
- Chagas, M.B.O.; Cordeiro, N.C.C.; Marques, K.M.R.; Pitta, M.G.R.; Rêgo, M.; Lima, M.C.A.; Pitta, M.G.R.; Pitta, I.R. New thiazacridine agents: Synthesis, physical and chemical characterization, and in vitro anticancer evaluation. Hum. Exp. Toxicol. 2017, 36, 1059–1070.
- Lafayette, E.A.; de Almeida, V.; Mônica, S.; Pitta, M.G.d.; Beltrão, E.I.C.; da Silva, T.G.; de Moura, R.O.; Pitta, I.d.; de Carvalho, L.B.; de Lima, M.D.C.A. Synthesis, DNA binding and topoisomerase I inhibition activity of thiazacridine and imidazacridine derivatives. Molecules 2013, 18, 15035–15050.
- Cui, Z.; Chen, S.; Wang, Y.; Gao, C.; Chen, Y.; Tan, C.; Jiang, Y. Design, synthesis and evaluation of azaacridine derivatives as dual-target EGFR and Src kinase inhibitors for antitumor treatment. Eur. J. Med. Chem. 2017, 136, 372–381.
- Karelou, M.; Kourafalos, V.; Tragomalou, A.P.; Marakos, P.; Pouli, N.; Tsitsilonis, O.E.; Gikas, E.; Kostakis, I.K. Synthesis, Biological Evaluation and Stability Studies of Some Novel Aza-Acridine Aminoderivatives. Molecules 2020, 25, 4584.